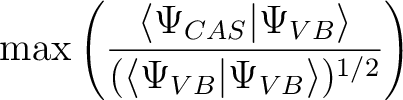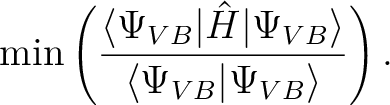MOLCAS manual:

Next: 8.5 ccsdt
Up: 8. Programs
Previous: 8.3 caspt2
Subsections
8.4 casvb
This program can be used in two basic modes:
| a) | variational optimization of quite general types of
nonorthogonal MCSCF or modern valence bond wavefunctions |
| b) | representation of CASSCF wavefunctions in modern valence form,
using overlap- (relatively inexpensive) or energy-based criteria.
|
For generating representations of CASSCF wavefunctions, the program
is invoked by the command CASVB.
For variational optimization of wavefunctions it is normally invoked
inside RASSCF by the sub-command VB (see ![[*]](crossref.png) ). ).
Bibliography: see [33,34,35,36].
8.4.1 Dependencies
The CASVB program needs the JOBIPH file from a RASSCF calculation,
and in addition also the ONEINT and ORDINT files from SEWARD.
8.4.2 Files
CASVB will use the following input
files: ONEINT, ORDINT, RUNFILE, JOBIPH,
(for more information see ), and
VBWFN with
valence bond wavefunction information (orbital and structure coefficients).
| File | Contents
|
| JOBIPH | On exit, the RASSCF interface file is overwritten with the
CASVB wavefunction.
|
| VBWFN | Valence bond wavefunction information (orbital and structure coefficients).
|
8.4.3 Input
This section describes the input to the CASVB program.
The input for each module is preceded by its name like:
&CASVB
8.4.3.1 Keywords
Optional keywords
| Keyword | Meaning
|
| END of Input | This marks the end of the input to the program.
|
Optional keywords to define the CASSCF wavefunction. Not generally required
because values stored in the job interface
file or used by the RASSCF program will normally be appropriate.
| Keyword | Meaning
|
| FROZen | Specifies frozen orbitals, as in the RASSCF program.
|
| INACtive | Specifies inactive orbitals, as in the RASSCF program.
|
| NACTel | Specifies the number of active electrons, as in the RASSCF program.
|
| RAS2 | Specifies RAS2 orbitals, as in the RASSCF program.
|
| SPIN | Specifies the total spin, as in the RASSCF program.
|
| SYMMetry | Specifies the CASSCF wavefunction symmetry, as in the RASSCF program.
|
Optional keywords to define the VB wavefunction
| Keyword | Meaning
|
| CON | The spatial
VB configurations are defined in terms of the active orbitals, and may be
specified using one or more CON keywords:
CON
n1 n2 n3 n4 ...
The configurations can be specified by occupation numbers, so that
ni is the occupation of the ith valence bond orbital. Alternatively a list of
Nact orbital numbers (in any order) may be provided – the
program determines which definition applies. The two specifications 1 0 1 2
and 1 3 4 4 are thus equivalent.
Input configurations are reordered by CASVB, so that configurations have
non-decreasing double occupancies. Configurations that are inconsistent with the
value for the total spin are ignored.
If no configurations are specified the single `covalent' configuration
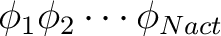 is assumed. is assumed.
|
| COUPle | COUPLE
key
key may be chosen from KOTANI (default), RUMER, PROJECT or LTRUMER,
specifying the scheme for constructing the
spin eigenfunctions used in the definition of valence bond structures. PROJECT
refers to spin functions generated using a spin projection operator, LTRUMER to
Rumer functions with the so-called ``leading term" phase convention.
|
| WAVE | WAVE
N S1 S2 ...
This keyword can be used to specify explicitly the number of electrons and spin(s) to
be used with a configuration list. If N is less than the present number of active electrons,
the input wavefunction fragment is assumed to form part of a direct product. Otherwise, the spins
specified may be greater than or equal to the SPIN value specified as input to the RASSCF
program. Defaults, for both N and S, are the values used by RASSCF.
|
Optional keywords for the recovery and/or storage of orbitals and vectors
| Keyword | Meaning
|
| STARt | START
key-1=filename-1
key-2=filename-2
...
Specifies input files for VB wavefunction (key-i = VB),
CASSCF CI vector (key-i = CI) and/or CASSCF molecular orbitals
(key-i = MO).
By default, the required information is taken from the file JOBOLD.
|
| SAVE | SAVE
key-1=filename-1
key-2=filename-2
...
Specifies output files for VB wavefunction (key-i = VB)
and/or the VB CI vector (key-i = VBCI). By default, the VB CI
vector is written to the file JOBIPH.
|
Optional keywords to override the starting guess
| Keyword | Meaning
|
| GUESs |
GUESS
key-1 ...
key-2 ...
ENDGUESs
The GUESS keyword initiates the input of a guess for the valence bond orbitals and/or
structure coefficients. key-i can be either ORB or STRUC.
These keywords
modify the guess provided by the program. It is
thus possible to modify individual orbitals in a previous solution
so as to construct the starting
guess. The ENDGUESs keyword terminates the guess input.
ORB
i c1 c2 ...cmact
Specifies a starting guess for valence bond orbital number i. The guess is specified
in terms of the mact active MOs defining the CASSCF wavefunction.
STRUC
c1 c2 ...cNVB
Specifies a starting guess for the NVB structure coefficients. If this keyword
is not provided, the perfect-pairing mode of
spin coupling is assumed for the spatial configuration having the least
number of doubly occupied orbitals.
Note that the definition of structures depends on the value of COUPLE. Doubly occupied
orbitals occur first in all configurations, and the spin eigenfunctions are based on the singly
occupied orbitals being in ascending order.
|
| ORBPerm | ORBPERM
i1 ...imact
Permutes the orbitals in the valence bond wavefunction and changes their phases according to
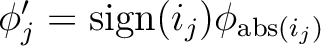 . The guess may be further modified using the
GUESS keyword. Additionally, the structure coefficients will be transformed
according to the given permutation (note that the configuration list must be closed under
the orbital permutation for this to be possible). . The guess may be further modified using the
GUESS keyword. Additionally, the structure coefficients will be transformed
according to the given permutation (note that the configuration list must be closed under
the orbital permutation for this to be possible).
|
Optional keywords for optimization control
| Keyword | Meaning
|
| CRIT | CRIT
method
Specifies the criterion for the optimization. method can be OVERLAP or ENERGY
(OVERLAP is default).
The former maximizes the normalized overlap with the CASSCF wavefunction:
and the latter simply minimizes the energy:
|
| MAXIter | MAXITER
Niter
Specifies the maximum number of iterations in the second-order optimizations. Default is Niter=50.
|
| (NO)CASProj |
(NO)CASPROJ
With this keyword the structure coefficients are picked from the transformed CASSCF CI vector, leaving
only the orbital variational parameters. For further details see the bibliography.
This option may be useful to aid convergence.
|
| SADDle | SADDLE
n
Defines optimization onto an n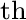 -order saddle point.
See also T. Thorsteinsson and D. L. Cooper, Int. J. Quant. Chem. 70, 637–50 (1998). -order saddle point.
See also T. Thorsteinsson and D. L. Cooper, Int. J. Quant. Chem. 70, 637–50 (1998).
|
| (NO)INIT | (NO)INIT
Requests a sequence of preliminary optimizations which aim to minimize the
computational cost while maximizing the likelihood of stable
convergence. This feature is the default if no wavefunction guess is available
and no OPTIM keyword specified in the input.
|
| METHod | METHOD
key
Selects the optimization algorithm to be used. key can be one
of: FLETCHER, TRIM, TRUSTOPT, DAVIDSON,
STEEP, VB2CAS, AUGHESS, AUG2,
CHECK, DFLETCH, NONE, or SUPER. Recommended are
the direct procedures DFLETCH or AUGHESS. For general
saddle-point optimization TRIM is used. Linear (CI only) optimization
problems use DAVIDSON. NONE suspends optimization, while
CHECK carries out a finite-difference check of the gradient and Hessian.
The default algorithm chosen by CASVB will be usually be adequate.
|
| TUNE | TUNE
...
Enables the input of individual parameters to be used in the optimization procedure
(e.g. for controlling step-size selection and convergence testing).
Details of the values used are output if print(3)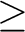 3 is specified.
For expert use only. 3 is specified.
For expert use only.
|
| OPTIm | More than one optimization may be performed in the same CASVB run,
by the use of OPTIM keywords:
OPTIM
[...
ENDOPTIM]
The subcommands may be any optimization declarations defined in this
section, as well as any symmetry or constraints specifications.
Commands given as arguments to OPTIM
will apply only to this optimization step, whereas commands specified
outside will act as default definitions for all subsequent OPTIM
specifications.
The OPTIM keyword
need not be specified if only one optimization step is required,
When only a machine-generated guess is available, CASVB will
attempt to
define a sequence of optimization steps that aims to maximize the
likelihood of successful convergence (while minimizing
CPU usage). To override this behaviour, simply specify one or more
OPTIM keywords. The ENDOPTIm keyword marks the end of the
specifications of an optimization step.
|
| ALTErn | A loop over two or more optimization steps may be specified using:
ALTERN
Niter
...
ENDALTERN
The program will repeat the specified optimization steps
until either all optimizations have converged, or the maximum iteration count,
Niter, has been reached.
The ENDALTErn keyword marks the end of the specification of an
ALTERN loop.
|
Optional keywords for definitions of molecular symmetry and any
constraints on the VB wavefunction
| Keyword | Meaning
|
| SYMElm | Various issues associated with symmetry-adapting valence bond wavefunctions
are considered, for example, in: T. Thorsteinsson, D. L. Cooper,
J. Gerratt and M. Raimondi, Theor. Chim. Acta 95, 131 (1997).
SYMELM
label sign
Initiates the definition of a symmetry operation referred to by label (any three characters).
sign can be + or -; it specifies whether the total wavefunction is symmetric or
antisymmetric under this operation, respectively. A value for sign is not always necessary
but, if provided, constraints will be put on the structure coefficients to ensure that the
wavefunction has the correct overall symmetry (note that the configuration list must be closed
under the orbital permutation induced by label for this to be possible).
The default for label is the identity.
The operator is defined in terms of its action on the active MOs as specified by
one or more of the keywords IRREPS, COEFFS, or TRANS. Any
other keyword, including optional use of the ENDSYMElm keyword, will
terminate the definition of this symmetry operator.
IRREPS
i1 i2 ...
The list i1 i2 ... specifies which irreducible representations (as defined in
the CASSCF wavefunction) are antisymmetric with respect to the label operation.
If an irreducible representation is not otherwise specified it is assumed to be symmetric
under the symmetry operation.
COEFFS
i1 i2 ...
The list i1 i2 ... specifies which individual CASSCF MOs are antisymmetric with
respect to the label operation. If an MO is not otherwise specified, it is assumed to be
symmetric under the symmetry operation. This specification may be useful if, for example, the
molecule possesses symmetry higher than that exploited in the CASSCF calculation.
TRANS
ndim i1 ...indim c11 c12
...c
Specifies a general n
dim x ndim transformation involving the MOs i1,
...indim,
specified by the c coefficients. This may be useful for systems with a two- or
three-dimensional irreducible representation, or if localized orbitals define the CASSCF
wavefunction. Note that the specified transformation must always be orthogonal.
|
| ORBRel | In general, for a VB wavefunction to be symmetry-pure, the orbitals must form a representation
(not necessarily irreducible) of the symmetry group. Relations between orbitals under
the symmetry operations defined by SYMELM may be specified according to:
ORBREL
i1 i2 label1 label2 ...
Orbital i1 is related to orbital i2 by the sequence of operations defined by the label
specifications (defined previously using SYMELM). The operators operate right to left. Note
that i1 and i2 may coincide. Only the minimum number of
relations required to define all the orbitals should be provided; an error exit
will occur if redundant ORBREL specifications are found.
|
| (NO)SYMProj | As an alternative to incorporating constraints, one may also ensure correct
symmetry of the wavefunction by use of a projection operator:
(NO)SYMPROJ
[irrep1 irrep2 ...]
The effect of this keyword is to set to zero the coefficients in unwanted
irreducible representations.
For this purpose, the symmetry group defined for the CASSCF wavefunction
is used (always a subgroup of D2h).
The list of irreps in the command specifies which components
of the wavefunction should be kept.
If no irreducible representations are given, the current
wavefunction symmetry is assumed. In a state-averaged calculation,
all irreps are retained for which a non-zero weight has been specified in the
wavefunction definition.
The SYMPROJ keyword may also be used in combination with constraints.
|
| FIXOrb | FIXORB
i1 i2 ...
This command freezes the orbitals specified in the list
i1 i2 ... to that of the starting guess. Alternatively the
special keywords ALL or NONE may be used. These orbitals
are eliminated from the optimization procedure, but will still be
normalized and symmetry-adapted according to any ORBREL
keywords given.
|
| FIXStruc | FIXSTRUC
i1 i2 ...
Freezes the coefficients for structures i1, i2,.... Alternatively
the special keywords ALL or NONE may be used. The
structures are eliminated from the optimization procedure, but may
still be affected by normalization or any symmetry keywords present.
|
| DELStruc | DELSTRUC
i1 i2,...
Deletes the specified structures from the wavefunction. The
special keywords ALL or NONE may be used. This specification should be compatible
with the other structure constraints present, as defined by SYMELM and ORBREL.
|
| ORTHcon | ORTHCON
key-1 ...
key-2 ...
...
The ORTHCON keyword initiates the input of orthogonality
constraints between pairs/groups of valence bond orbitals.
The sub-keywords key-i can be any of ORTH, PAIRS,
GROUP, STRONG or FULL. Orthogonality constraints
should be used with discretion. Note that orthogonality constraints
for an orbital generated from another by symmetry operations (using the
ORBREL keyword) cannot in general be satisfied. The ENDORTHcon
keyword can be used to terminate the input of orthogonality constraints.
ORTH
i1 i2 ...
Specifies a list of orbitals to be orthogonalized. All overlaps
between pairs of orbitals in the list are set to zero.
PAIRS i1 i2 ...
Specifies a simple list of orthogonalization pairs. Orbital i1 is
made orthogonal to i2, i3 to i4, etc.
GROUP label i1 i2 ...
Defines an orbital group to be used with the ORTH or
PAIRS keyword. The group is referred to by label which
can be any three characters beginning with a letter a–z. Labels
defining different groups can be used together or in combination
with orbital numbers in ORTH or PAIRS.
i1 i2 ... specifies
the list of orbitals in the group. Thus the combination
GROUP AAA 1 2 GROUP BBB 3 4 ORTH AAA BBB will orthogonalize
the pairs of orbitals 1-3, 1-4, 2-3 and 2-4.
STRONG
This keyword is short-hand for strong orthogonality. The only allowed
non-zero overlaps are between pairs of orbitals (2n-1, 2n).
FULL
This keyword is short-hand for full orthogonality and is mainly
useful for testing purposes.
|
Optional keywords for wavefunction analysis
| Keyword | Meaning
|
| CIWEights | For further details regarding the calculation of weights in CASVB, see
T. Thorsteinsson and D. L. Cooper, J. Math. Chem. 23, 105-26 (1998).
CIWEIGHTS
key1 key2 ...[ ] ]
Prints weights of the CASSCF wavefunction transformed
to the basis of nonorthogonal VB structures. For the key options
see VBWEIGHTS below. Note that the evaluation of inverse overlap
weights involves an extensive computational overhead for large active
spaces. Weights are given for the
total CASSCF wavefunction, as well as the orthogonal complement to
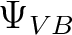 . The default for the number of configurations requested,
, is 10. If =-1 all configurations are
included. . The default for the number of configurations requested,
, is 10. If =-1 all configurations are
included.
|
| REPOrt | REPORT
[...
ENDREPORT]
Outputs orbital/structure coefficients and derived information.
The ENDREPOrt keyword can be used to mark the end of the specification
of a report step.
|
| (NO)SCORr | (NO)SCORR
With this option, expectation values of the spin operators
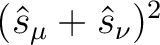 are evaluated for all pairs of are evaluated for all pairs of 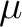 and and
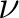 . Default is NOSCORR. The procedure is described by: G. Raos,
J. Gerratt, D. L. Cooper and M. Raimondi,
Chem. Phys. 186, 233–250 (1994); ibid, 251–273 (1994);
D. L. Cooper, R. Ponec, T. Thorsteinsson and G. Raos,
Int. J. Quant. Chem. 57, 501–518 (1996). . Default is NOSCORR. The procedure is described by: G. Raos,
J. Gerratt, D. L. Cooper and M. Raimondi,
Chem. Phys. 186, 233–250 (1994); ibid, 251–273 (1994);
D. L. Cooper, R. Ponec, T. Thorsteinsson and G. Raos,
Int. J. Quant. Chem. 57, 501–518 (1996).
This analysis is currently only implemented for spin-coupled wavefunctions.
|
| VBWEights | For further details regarding the calculation of weights in CASVB, see
T. Thorsteinsson and D. L. Cooper, J. Math. Chem. 23, 105-26 (1998).
VBWEIGHTS
key1 key2 ...
Calculates and outputs weights of the structures in the valence bond
wavefunction . key specifies the definition of
nonorthogonal weights to be used, and can be one of:
- CHIRGWIN
- Evaluates Chirgwin-Coulson weights (see:
B. H. Chirgwin and C. A. Coulson, Proc. Roy. Soc. Lond. A201,
196 (1950)).
- LOWDIN
- Performs a symmetric orthogonalization of the
structures and outputs the subsequent weights.
- INVERSE
- Outputs ``inverse overlap populations" as in
G. A. Gallup and J. M. Norbeck, Chem. Phys. Lett. 21, 495–500 (1973).
- ALL
- All of the above.
- NONE
- Suspends calculation of structure weights.
The commands LOWDIN and INVERSE require the overlap matrix
between valence bond structures, so that some additional computational
overhead is involved.
|
Optional keywords for further general options
| Keyword | Meaning
|
| PREC | PREC
iprec iwidth
Adjusts the precision for printed quantities. In most cases, iprec simply refers
to the number of significant digits after the decimal point. Default is iprec=+8.
iwidth specifics the maximum width of printed output, used when determining
the format for printing arrays.
|
| PRINt | PRINT
i1 i2 ...
Each number specifies the level of output required at various stages of the execution, according to the
following convention:
- -1
- No output except serious, or fatal, error messages.
- 0
- Minimal output.
- 1
- Standard level of output.
- 2
- Extra output.
The areas for which output can be controlled are:
- i1
- Print of input parameters, wavefunction definitions, etc.
- i2
- Print of information associated with symmetry constraints.
- i3
- General convergence progress.
- i4
- Progress of the 2nd-order optimization procedure.
- i5
- Print of converged solution and analysis.
- i6
- Progress of variational optimization.
- i7
- File usage.
For all, the default output level is +1. If i52 VB orbitals will
be printed in the AO basis (provided that the definition of MOs is
available).
|
| SHSTruc | Prints overlap and Hamiltonian matrices between VB structures.
|
| STATs | STATS
Prints timing and usage statistics.
|
&seward
symmetry
x y
basis set
c.sto-3g....
c 0 0 -0.190085345
end of basis
basis set
h.sto-3g....
h 0 1.645045225 1.132564974
end of basis
&scf
occupied
3 0 1 0
&rasscf
inactive
1 0 0 0
ras2
3 1 2 0
nactel
6 0 0
lumorb
&casvb
In many cases it can be helpful to view the shape of the converged valence bond orbitals, and
Molcas therefore provides two facilities for doing this. For the Molden program, an interface file
is generated at the end of each CASVB run (see also Section ).
Alternatively a CASVB run may be followed by RASSCF to get orbitals
(Section ) and GRID_IT with the VB specification
(Section ), in order to generate a three-dimensional grid, for viewing, for example,
with LUSCUS program.
Next: 8.5 ccsdt
Up: 8. Programs
Previous: 8.3 caspt2
|