




















MOLCAS manual: Next: 8.42 rassi Up: 8. Programs Previous: 8.40 quater
|
 -vectors in the
Davidson procedure and to compute the two-body density matrices.
The upper limit to the size of the CASSCF wave function that can be
handled with the present program is about 107 CSFs and is,
in general, limited by the dynamic work array available to the
program.
-vectors in the
Davidson procedure and to compute the two-body density matrices.
The upper limit to the size of the CASSCF wave function that can be
handled with the present program is about 107 CSFs and is,
in general, limited by the dynamic work array available to the
program.
 and
and  orbitals will then appear in irrep 1. If the input
orbitals have been prepared to be adapted to linear symmetry, the
Supsym input can be used to keep this symmetry through the iterations.
The program will do this automatically with the use of the
input keyword LINEAR. Similarly, for single atoms, spherical
symmetry can be enforced by the keyword ATOM.
orbitals will then appear in irrep 1. If the input
orbitals have been prepared to be adapted to linear symmetry, the
Supsym input can be used to keep this symmetry through the iterations.
The program will do this automatically with the use of the
input keyword LINEAR. Similarly, for single atoms, spherical
symmetry can be enforced by the keyword ATOM.
| File | Contents |
| JOBIPH | This file is written in binary format and carries the results of the wave function optimization such as MO- and CI-coefficients. If several consecutive RASSCF calculations are made, the file names will be modified by appending '01','02' etc. |
| RUNFILE | The RUNFILE is updated with information from the RASSCF calculation such as the first order density and the Fock matrix. |
| MD_CAS.x | Molden input file for molecular orbital analysis for CI root x. |
| RASORB | This ASCII file contains molecular orbitals, occupation numbers, and orbital indices from a RASSCF calculation. The natural orbitals of individual states in an average-state calculation are also produced, and are named RASORB.1, RASORB.2, etc. |
| MCDENS | This ASCII file is generated for MC-PDFT calculations. It contains spin densities, total density and on-top pair density values on grid (coordinates in a.u.). |
8.41.5 Input
This section describes the input to the
RASSCF program in the MOLCAS program system. The input starts
with the program name
&RASSCF
There are no compulsory keywords, but almost any meaningful calculation will require some keyword. At the same time, most choices have default settings, and many are able to take relevant values from earlier calculations, from available orbital files, etc.
To run an MC-PDFT calculation in the RASSCF module, the keywords CIONLY, KSDFT, ROKS and the functional choice are needed. The currently available functionals are tPBE, tBLYP and tLSDA. Also: LUMORB is needed if external orbitals are used. JOBIPH is needed if external orbital stored in JobIph files are used. CIRESTART is needed if a pre-optimized CI vector stored in JOBIPH is to be used.
8.41.5.1 Optional keywords
There is a large number of optional keywords you can specify. They are used to specify the orbital spaces, the CI wave function etc., but also more arcane technical details that can modify e.g. the convergence or precision. The first 4 characters of the keyword are recognized by the input parser and the rest is ignored. If not otherwise stated the numerical input that follows a keyword is read in free format. A list of these keywords is given below:
| Keyword | Meaning | ||||||||||
| TITLe | Follows the title for the calculation in a single line | ||||||||||
| SYMMetry | Specify the selected symmetry type (the irrep) of the wave function as a number between 1 and 8 (see SYMMETRY keyword in GATEWAY section). Default is 1, which always denote the totally symmetric irrep. | ||||||||||
| SPIN | The keyword is followed by an integer giving the value of spin multiplicity (2S+1). Default is 1 (singlet). | ||||||||||
| CHARge | Specify the total charge on the system as an integer. If this keyword is used, the NACTEL keyword should not be used, unless the symmetry group is C1 and INACTIVE is not used (in this case the number of inactive orbitals will be computed from the total charge and active electrons). Default value: 0 | ||||||||||
| RASScf | Specify two numbers: maximum number of holes in RAS1 and the maximum number of electrons occupying the RAS3 orbitals Default values are: 0,0 See also keyword CHARGE and NACTEL. The specification using RASSCF, and CHARGE if needed, together replace the single keyword NACTEL. | ||||||||||
| NACTel | Requires one or three numbers to follow, specifying
| ||||||||||
| CIROot | Specifies the CI root(s) and the dimension of
the starting CI matrix used in the CI Davidson procedure. This input
makes it possible to perform orbital optimization for the average
energy of a number of states. The first line of input gives two or three
numbers, specifying the number of roots used in the average
calculation (NROOTS), the dimension of the small CI matrix in
the Davidson procedure (LROOTS), and possibly a non-zero integer IALL.
If IALL.ne.1 or there is no IALL, the second line gives the index of
the states over which the average is taken (NROOTS numbers,
IROOT). Note that the size of the CI matrix, LROOTS, must be at least as
large as the highest root, IROOT. If, and only if, NROOTS>1 a third
line follows, specifying the weights of the different states in the average
energy. If IALL=1 has been specified, no more lines are read. A state average
calculation will be performed over the NROOTS lowest states with equal weights.
energy. Examples:
CIRoot= 3 5; 2 4 5; 1 1 3 The average is taken over three states corresponding to roots 2, 4, and 5 with weights 20%, 20%, and 60%, respectively. The size of the Davidson Hamiltonian is 5. Another example is: CIRoot= 19 19 1 A state average calculation will be performed over the 19 lowest states each with the weight 1/19 Default values are NROOTS = LROOTS = IROOT = 1 (ground state), which is the same as the input: CIRoot= 1 1; 1 | ||||||||||
| CISElect | This keyword is used to select CI roots by an overlap criterion. The input consists of three lines per root that is used in the CI diagonalization (3*NROOTS lines in total). The first line gives the number of configurations used in the comparison, nRef, up to five. The second line gives nRef reference configuration indices. The third line gives estimates of CI coefficients for these CSF's. The program will select the roots which have the largest overlap with this input. Be careful to use a large enough value for LROOTS (see above) to cover the roots of interest. | ||||||||||
| ATOM | This keyword is used to get orbitals with pure spherical symmetry for atomic calculations (the radial dependence can vary for different irreps though). It causes super-symmetry to be switched on (see SUPSym keyword) and generates automatically the super-symmetry vector needed. Also, at start and after each iteration, it sets to zero any CMO coefficients with the wrong symmetry. Use this keyword instead of SUPSym for atoms. | ||||||||||
| LINEar | This keyword is used to get orbitals with pure rotational symmetry for linear molecules. It causes super-symmetry to be switched on (see SUPSym keyword) and generates automatically the super-symmetry vector needed. Also, at start and after each iteration, it sets to zero any CMO coefficients with the wrong symmetry. Use this keyword instead of SUPSym for linear molecules. | ||||||||||
| RLXRoot | Specifies which root to be relaxed in a geometry optimization of a
state average wave function. Thus, the keyword has to be combined
with CIRO.
In a geometry optimization the following input
CIRoot= 3 5; 2 4 5; 1 1 3 RLXRoot= 4 will relax CI root number 4. | ||||||||||
| MDRLxroot | Selects a root from a state average wave function for gradient computation in the first step of a molecular dynamics simulation. The root is specified in the same way as in the RLXR keyword. In the following steps the trajectory surface hopping can change the root if transitions between the states occur. This keyword is mutually exclusive with the RLXR keyword. | ||||||||||
| EXPErt | This keyword forces the program to obey the input. Normally, the program can decide to change the input requests, in order to optimize the calculation. Using the EXPERT keyword, such changes are disallowed. | ||||||||||
| RFPErt | This keyword will add a constant reaction field perturbation to the Hamiltonian. The perturbation is read from the RUNOLD (if not present defults to RUNFILE) and is the latest self-consistent perturbation generated by one of the programs SCF or RASSCF. | ||||||||||
| NONEquilibrium | Makes the slow components of the reaction field of another state present in the reaction field calculation (so-called non-equilibrium solvation). The slow component is always generated and stored on file for equilibrium solvation calculations so that it potentially can be used in subsequent non-equilibrium calculations on other states. | ||||||||||
| RFROot | Enter the index of that particular root in a state-average calculation for which the reaction-field is generated. It is used with the PCM model. | ||||||||||
| CIRFroot | Enter the relative index of one of the roots specified in CISElect for which the reaction-field is generated. Used with the PCM model. | ||||||||||
| NEWIph | The default name of the JOBIPH file will be determined by any already existing such files in the work directory, by appending '01', '02' etc. so a new unique name is obtained. | ||||||||||
| FROZen | Specifies the number of frozen orbitals in each symmetry. (see below for condition on input orbitals). Frozen orbitals will not be modified in the calculation. Only doubly occupied orbitals can be left frozen. This input can be used for example for inner shells of heavy atoms to reduce the basis set superposition error. Default is 0 in all symmetries. | ||||||||||
| INACtive | Specify on the next line the number of inactive (doubly occupied) orbitals in each symmetry. Frozen orbitals should not be included here. Default is 0 in all symmetries, but if there is no symmetry (C1) and both CHARGE and NACTEL are given, the number of inactive orbitals will be calculated automatically. | ||||||||||
| RAS1 | On the next line specify the number of orbitals in each symmetry for the RAS1 orbital subspace. Default is 0 in all symmetries. | ||||||||||
| RAS2 | On the next line specify the number of orbitals in each symmetry for the RAS2 orbital subspace. Default is 0 in all symmetries. | ||||||||||
| RAS3 | On the next line specify the number of orbitals in each symmetry for the RAS3 orbital subspace. Default is 0 in all symmetries. | ||||||||||
| DELEted | Specify the number of deleted orbitals in each symmetry. These orbitals will not be allowed to mix into the occupied orbitals. It is always the last orbitals in each symmetry that are deleted. Default is 0 in all symmetries, unless orbitals wer already deleted by previous programs due to near-linear dependence. | ||||||||||
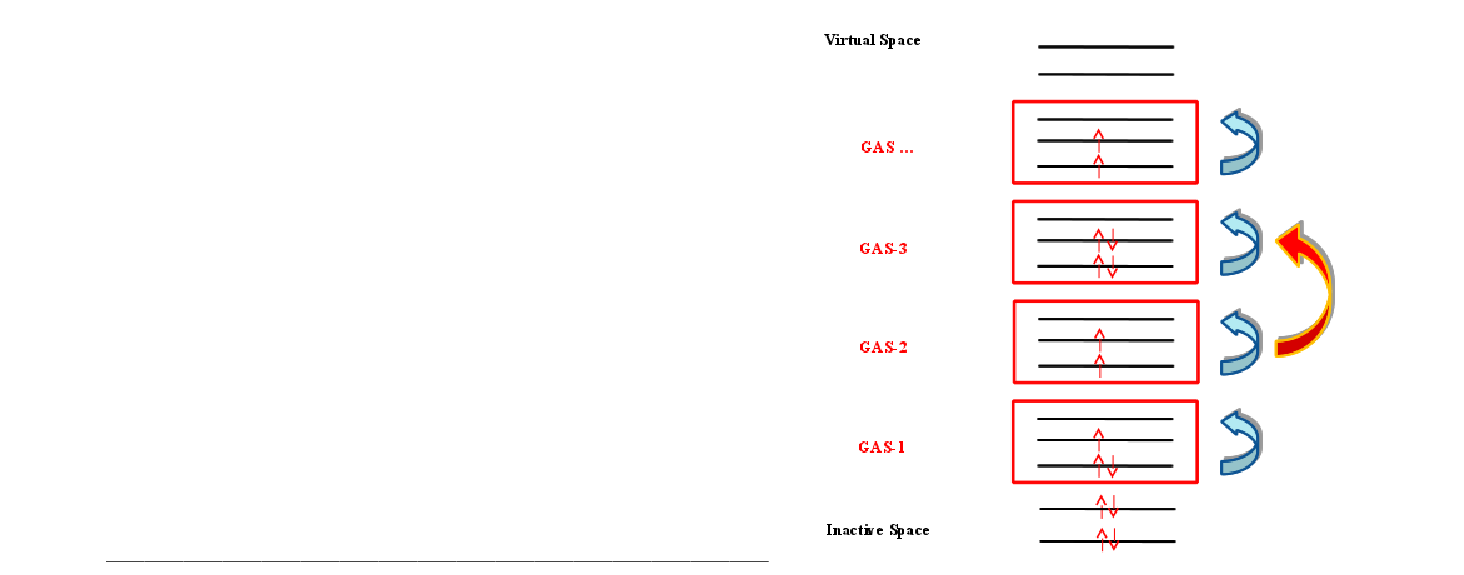
| GASScf | Needed to perform a Generalized Active Space (GASSCF) calculation.
It is followed by an integer that defines the number of active subspaces,
and two lines for each subspace. The first line gives the number of orbitals
in each symmetry, the second gives the minimum and maximum number of
electrons in the accumulated active space.
An example of an input that uses this keyword is the following:
In the example above (20in32), excitations from one subspace to another are not allowed since the values of MIN and MAX for GSOC are identical for each of the five subspaces. | ||||||||||
| KSDFT | Needed to perform MC-PDFT calculations. It must be used together with
CIONLY keyword (it is a post-SCF method not compatible with SCF) and ROKS keyword.
The functional choice follows. Currently available functionals are: tPBE, tBLYP, tLSDA.
An example of an input that uses this keyword follows:
&RASSCF JOBIPH CIRESTART CIONLY Ras2 1 0 0 0 1 0 0 0 KSDFT ROKS TPBE In the above example, JOBIPH is used to use orbitals stored in JobIph, CIRESTART is used to use a pre-optimized CI vector, CIONLY is used to avoid conflicts between the standard RASSCF module and the MC-PDFT method (not compatible with SCF so far). The functional chosen is the translated–PBE. | ||||||||||
| JOBIph | Input molecular orbitals are read from an unformatted file with FORTRAN file name JOBOLD. Note, the keywords Lumorb, Core, and Jobiph are mutually exclusive. If none is given the program will search for input orbitals on the runfile in the order: RASSCF, SCF, GUESSORB. If none is found, the keyword CORE will be activated. | ||||||||||
| IPHName | Override the default choice of name of the JOBIPH file by giving the file name you want. The name will be truncated to 8 characters and converted to uppercase. | ||||||||||
| LUMOrb | Input molecular orbitals are read from a formatted file with FORTRAN file name INPORB. Note, the keywords Lumorb, Core, and Jobiph are mutually exclusive. If none is given the program will search for input orbitals on the runfile in the order: RASSCF, SCF, GUESSORB. If none is found, the keyword CORE will be activated. | ||||||||||
| FILEorb | Override the default name (INPORB) for starting orbital file by giving the file name you want. | ||||||||||
| CORE | Input molecular orbitals are obtained by diagonalizing the core Hamiltonian. This option is only recommended in simple cases. It often leads to divergence. Note, the keywords Lumorb, Core, and Jobiph are mutually exclusive. | ||||||||||
| ALPHaOrBeta | With UHF orbitals as input, select alpha or beta as starting orbitals. A positive value selects alpha, a negative value selects beta. Default is 0, which fails with UHF orbitals. This keyword does not affect the spin of the wave function (see the SPIN keyword). | ||||||||||
| TYPEIndex | This keyword forces the program to use information about subspaces from the
INPORB file.
User can change the order of orbitals by editing of "Type Index" section in the INPORB file. The legend of the types is:
| ||||||||||
| ALTEr | This keyword is used to change the ordering of MO in INPORB or
JOBOLD. The keyword requires first the number of pairs to be interchanged,
followed, for each pair, the symmetry species of
the pair and the indices of the two permuting MOs. Here is an example:
ALTEr= 2; 1 4 5; 3 6 8 In this example, 2 pairs of MO will be exchanged: 4 and 5 in symmetry 1 and 6 and 8 in symmetry 3. | ||||||||||
| CLEAnup | This input is used to set to zero specific coefficients of the input
orbitals. It is of value, for example, when the actual symmetry is
higher than given by input and the trial orbitals are contaminated
by lower symmetry mixing. The input requires at least one line
per symmetry specifying the number of additional groups of orbitals
to clean. For each group of orbitals within the symmetry, three lines
follow. The first line indicates the number of considered orbitals
and the specific number of the orbital (within the symmetry) in the
set of input orbitals. Note the input lines can not be longer than 72
characters and the program expects as many continuation lines as are
needed. The second line indicates the number of
coefficients belonging to the prior orbitals which are going to be
set to zero and which coefficients. The third line indicates the
number of the coefficients of all the complementary orbitals of
the symmetry which are going to be set to zero and which are these
coefficients. Here is an example of what an input would look like:
CLEAnup 2 3 4 7 9; 3 10 11 13; 4 12 15 16 17 2 8 11; 1 15; 0 0; 0; 0 In this example the first entry indicates that two groups of orbitals are specified in the first symmetry. The first item of the following entry indicates that there are three orbitals considered (4, 7, and 9). The first item of the following entry indicates that there are three coefficients of the orbitals 4, 7, and 9 to be set to zero, coefficients 10, 11, and 13. The first item of the following entry indicates that there are four coefficients (12, 15, 16, and 17) which will be zero in all the remaining orbitals of the symmetry. For the second group of the first symmetry orbitals 8 and 11 will have their coefficient 15 set to zero, while nothing will be applied in the remaining orbitals. If a geometry optimization is performed the keyword is inactive after the first structure iteration. | ||||||||||
| CIREstart | Starting CI-coefficients are read from a binary file JOBOLD. | ||||||||||
| ORBOnly | This input keyword is used to get a formated ASCII file (RASORB, RASORB.2, etc) containing molecular orbitals and occupations reading from a binary JobIph file. The program will not perform any other operation. (In this usage, the program can be run without any files, except the JOBIPH file). | ||||||||||
| CIONly | This keyword is used to disable orbital optimization, that is, the CI roots are computed only for a given set of input orbitals. | ||||||||||
| CHOInput | This marks the start of an input section for modifying
the default settings of the Cholesky RASSCF.
Below follows a description of the associated options.
The options may be given in any order,
and they are all optional except for
ENDChoinput which marks the end of the CHOInput section.
| ||||||||||
| OFEMbedding | Performs a Orbital-Free Embedding (OFE)RASSCF calculation, available only in combination with Cholesky or RI integral representation.
The runfile of the environment subsystem renamed AUXRFIL is required.
An example of input for the keyword OFEM is the following:
OFEMbedding ldtf/pbe dFMD 1.0 1.0d2 FTHAw 1.0d-4 The keyword OFEM requires the specification of two functionals in the form fun1/fun2, where fun1 is the functional used for the Kinetic Energy (available functionals: Thomas-Fermi, with acronym LDTF, and the NDSD functional), and where fun2 is the xc-functional (LDA, LDA5, PBE and BLYP available at the moment). The OPTIONAL keyword dFMD has two arguments: first, the fraction of correlation potential to be added to the OFE potential; second, the exponential decay factor for this correction (used in PES calculations). The OPTIONAL keyword dFMD specifies the fraction of correlation potential to be added to the OFE potential. The OPTIONAL keyword FTHA is used in a freeze-and-thaw cycle (EMIL Do While) to specify the (subsystems) energy convergence threshold. | ||||||||||
| ITERations | Specify the maximum number of RASSCF iterations, and the maximum number of iterations used in the orbital optimization (super-CI) section. Default and maximum values are 200,100. | ||||||||||
| LEVShft | Define a level shift value for the super-CI Hamiltonian. Typical values are in the range 0.0 – 1.5. Increase this value if a calculation diverges. The default value 0.5, is normally the best choice when Quasi-Newton is performed. | ||||||||||
| THRS | Specify convergence thresholds for: energy, orbital rotation matrix, and energy gradient. Default values are: 1.0e-08, 1.0e-04, 1.0e-04. | ||||||||||
| TIGHt | Convergence thresholds for the Davidson diagonalization procedure. Two numbers should be given: THREN and THFACT. THREN specifies the energy threshold in the first iteration. THFACT is used to compute the threshold in subsequent iterations as THFACT*DE, where DE is the RASSCF energy change. Default values are 1.0d-04 and 1.0d-3. | ||||||||||
| NOQUne | This input keyword is used to switch off the Quasi-Newton update procedure for the Hessian. Pure super-CI iterations will be performed. (Default setting: QN update is used unless the calculation involves numerically integrated DFT contributions.) | ||||||||||
| QUNE | This input keyword is used to switch on the Quasi-Newton update procedure for the Hessian. (Default setting: QN update is used unless the calculation involves numerically integrated DFT contributions.) | ||||||||||
| CIMX | Specify the maximum number of iterations allowed in the CI procedure. Default is 100 with maximum value 200. | ||||||||||
| HEXS | Highly excited states. Will eliminate the maximum occupation in one or more RAS/GAS's thereby eliminating all roots below. Very helpful for core excitations where the ground-state input can be used to eliminate unwanted roots. Works with RASSI. First input is the number of RAS/GAS where the maximum occupation should be eliminated. Second is the RAS/GAS or RAS/GAS's where maximum occupation will not be allowed. | ||||||||||
| SDAV | Here follows the dimension of the explicit Hamiltonian used to speed up the Davidson CI iteration process. An explicit H matrix is constructed for those configurations that have the lowest diagonal elements. This H-matrix is used instead of the corresponding diagonal elements in the Davidson update vector construction. The result is a large saving in the number if CI iterations needed. Default value is the smallest of 100 and the number of configurations. Increase this value if there is problems converging to the right roots. | ||||||||||
| SXDAmp | A variable called SXDAMP regulates the size of the orbital rotations. Use keyword SXDAmp and enter a real number. The default value is 0.0002. Larger values can give slow convergence, very low values may give problems e.g. if some active occupations are very close to 0 or 2. | ||||||||||
| SUPSym | This input is used to restrict possible orbital
rotations. It is of value, for example, when the actual symmetry is
higher than given by input. Each orbital is given a label IXSYM(I).
If two orbitals in the same symmetry have different labels they will
not be allowed to rotate into each other and thus prevents from obtaining
symmetry broken solutions. Note, however, that the starting orbitals must
have the right symmetry. The input requires one or more entries
per symmetry. The first specifies the number of additional subgroups in this
symmetry ( a 0 (zero) denotes that there is no additional subgroups and the
value of IXSYM will be 0 (zero) for all orbitals in that symmetry ).
If the number of additional subgroups is not zero there are additional
entries for each subgroup: The dimension of the subgroup and
the list of orbitals in the subgroup counted relative to the first orbital
in this symmetry. Note, the input lines can not be longer than 180 characters
and the program expects continuation lines as many as there are needed.
As an example assume an atom treated in C2v symmetry for
which the dz2 orbitals (7 and 10) in symmetries 1 may mix with the
s orbitals. In addition, the dz2 and dx2-y2 orbitals (8 and 11)
may also mix. Then the input would look like:
SUPSym 2 2 7 10; 2 8 11 0; 0; 0 In this example the first entry indicates that we would like to specify two additional subgroups in the first symmetry (total symmetric group). The first item in the following two entries declares that each subgroup consists of two orbitals. Orbitals 7 and 10 constitute the first group and it is assumed that these are orbitals of dz2 character. The second group includes the dx2-y2 orbitals 8 and 11. The following three entries denote that there are no further subgroups defined for the remaining symmetries. Ordering of the orbitals according to energy is deactivated when using SUPSym. If you activate ordering using ORDEr, the new labels will be printed in the output section. If a geometry optimization is performed the reordered matrix will be stored in the RUNFILE file and read from there instead of from the input in each new structure iteration. | ||||||||||
| HOME | With this keyword, the root selection in the Super-CI orbital update is by maximum overlap rather than lowest energy. | ||||||||||
| IVO | The RASSCF program will diagonalize the core Hamiltonian in the space of virtual orbitals, before printing them in the output. The resulting orbitals are only suitable to select which one should enter the active space in a subsequent calculation. The RASSCF calculation is not suitable for CASPT2/MRCI or any other correlated methods, becasue the energies of the virtual orbitals is undefined. This keyword is equivalent to IVO keyword of the SCF program. | ||||||||||
| VB | Using this keyword, the CI optimization step in the RASSCF program will be
replaced by a call to the CASVB program, such that fully variational valence
bond calculations may be carried out. The VB keyword can be followed by any
of the directives described in section ![[*]](crossref.png) and should be terminated
by ENDVB. Energy-based optimization of the VB parameters is the default,
and the output level for the main CASVB iterations is reduced to -1,
unless the print level for CASVB print option 6 is and should be terminated
by ENDVB. Energy-based optimization of the VB parameters is the default,
and the output level for the main CASVB iterations is reduced to -1,
unless the print level for CASVB print option 6 is 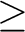 2. 2.
| ||||||||||
The keyword is followed by a line giving the print
levels for various logical code sections. It has the following structure:
IW IPR IPRSEC(I), I=1,7
Print= 6 2 2 2 3 2 2 2 2 | |||||||||||
| MAXOrb | Maximum number of RasOrb files to produce, one for each root up to the maximum. | ||||||||||
| OUTOrbitals | This keyword is used to select the type of orbitals to be written in a formated ASCII file. By default a formated RASORB file containing average orbitals and subsequent RASORB.1, RASORB.2, etc, files containing natural orbitals for each of the computed (up to ten) roots will be generated in the working directory. An entry follows with an additional keyword selecting the orbital type. The possibilities are: AVERage orbitals: this is the default option. This keyword is used to produce a formated ASCII file of orbitals (RASORB) which correspond to the final state average density matrix obtained by the RASSCF program. The inactive and secondary orbitals have been transformed to make an effective Fock matrix diagonal. Corresponding diagonal elements are given as orbital energies in the RASSCF output listing. The active orbitals have been obtained by diagonalizing the sub-blocks of the average density matrix corresponding to the three different RAS orbital spaces, thereby the name pseudo-natural orbitals. They will be true natural orbitals only for a CASSCF wave function. CANOnical orbitals: Use this keyword to produce the canonical orbitals. They differ from the natural orbitals, because also the active part of the Fock matrix is diagonalized. Note that the density matrix is no longer diagonal and the CI coefficients have not been transformed to this basis. This option substitutes the previous keyword CANOnical. NATUral orbitals: Use this keyword to produce the true natural orbitals. The keyword should be followed by a new line with an integer specifying the maximum CI root for which the orbitals and occupation numbers are needed. One file for each root will be generated up to the specified number. In a one state RASSCF calculation this number is always 1, but if an average calculation has been performed, the NO's can be obtained for all the states included in the energy averaging. Note that the natural orbitals main use is as input for property calculations using SEWARD. The files will be named RASORB, RASORB.2, RASORB.3, etc. This keyword is on by default for up to ten roots. SPIN orbitals. This keyword is used to produce a set of spin orbitals and is followed by a new line with an integer specifying the maximum CI root for which the orbitals and occupation numbers are needed. One file for each root will be generated up to the specified number. Note, for convenience the doubly occupied and secondary orbitals have been added to these sets with occupation numbers 0 (zero). The main use of these orbitals is to act as input to property calculations and for graphical presentations. This keyword is on by default for up to ten roots.
An example input follows in which five files are requested containing
natural orbitals for roots one to five of a RASSCF calculation.
The files are named RASORB.1, RASORB.2, RASORB.3, RASORB.4, and RASORB.5,
respectively for each one of the roots.
Although this is the default, it can be used complemented by the ORBOnly
keyword, and the orbitals will be read from
a JobIph file from a previous calculation, in case the formated files
were lost or you require more than ten roots. As an option the
MAXOrb can be also used to increase the number of files
over ten.
| ||||||||||
| ORBListing | This keyword is followed with a word showing
how extensive you want the orbital listing to be in the printed output.
The alternatives are:
| ||||||||||
| ORBAppear | This keyword requires an entry with a word showing
the appearance of the orbital listing in the printed output.
The alternatives are:
| ||||||||||
| PROR | This keyword is used to alter the printout of the MO-coefficients. Two numbers must be given of which the first is an upper boundary for the orbital energies and the second is a lower boundary for the occupation numbers. Orbitals with energy higher than the threshold or occupation numbers lower that the threshold will not be printed. By default these values are set such that all occupied orbitals are printed, and virtual orbitals with energy less than 0.15 au. However, the values are also affected by use of OUTPUT. | ||||||||||
| PRSD | This keyword is used to request that not only CSFs are printed with the CI coefficients, but also the determinant expansion. | ||||||||||
| ORDEr | This input keyword is used to deactivate or activate ordering of the output orbitals according to energy. One number must be given: 1 if you want ordering and 0 if you want to deactivate ordering. Default is 1 and with SUPSym keyword default is 0. | ||||||||||
| PRSP | Use this keyword to get the spin density matrix for the active orbitals printed. | ||||||||||
| PRWF | Enter the threshold for CI coefficients to be printed (Default: 0.05). | ||||||||||
| TDM | If this keyword is given, and if HDF5 support is enabled, the active 1-electron transition density matrix between every pair of states in the current calculation will be computed and stored in the HDF5 file. | ||||||||||
| DMRG | Specify maximum number of renormalized states (or virtual bond dimension m) in each microiteration in DMRG calculations. m must be integer and should be at least 500. This keyword is supported in both CheMPS2 and Block interfaces. Note that DMRG-CASSCF calculations for excited states are not fully supported by the Block interface. | ||||||||||
| 3RDM | Use this keyword to get the 3-particle and Fock matrix contracted with the 4-particle reduced density matrices (3-RDM and F.4-RDM) for DMRG-CASPT2. OUTOrbitals = CANOnical is automatically activated. In CheMPS2 interface, both 3-RDM and F.4-RDM are calculated. In Block interface, only 3-RDM is calculated while F.4-RDM is approximated in the CASPT2 module. | ||||||||||
| CHBLb | Specify a threshold for activating restart in CheMPS2. After each macroiteration, if the max BLB value is smaller than CHBLb, activate partial restart in CheMPS2. If the max BLB value is smaller than CHBLb/10.0, activate full restart in CheMPS2. Default value is: 0.5d-2. | ||||||||||
| DAVTolerance | Specify value for Davidson tolerance in CheMPS2. Default value is 1.0d-7. | ||||||||||
| NOISe | Specify value for noise pre-factor in CheMPS2. This noise is set to 0.0 in the last instruction. Default value (recommended) is: 0.05 | ||||||||||
| MXSWeep | Maximum number of sweeps in the last instruction in CheMPS2. Default value is: 8. In the last iteration of DMRG-SCF, MXSW is increased by five times (default 40). | ||||||||||
| MXCAnonical | Maximum number of sweeps in the last instruction with pseudocanonical orbitals in CheMPS2. Default value is: 40. | ||||||||||
| CHREstart | Use this keyword to activate restart in the first DMRG iteration from a previous calculation. The working directory must contain molcas_natorb_fiedler.txt and CheMPS2_natorb_MPSx.h5 (x=0 for the ground state, 1 for the first excited state, etc). If these files are not in the working directory, a warning is printed at the beginning of the calculation and restart is skipped (start from scratch). | ||||||||||
| DMREstart | Use this keyword to activate restart in the last DMRG iteration from the previous iteration or calculation.
This keyword only works when using OUTOrbitals = CANOnical or 3RDM.
DMREstart = 0 (default): start from scratch to calculate 3-RDM and F.4-RDM. DMREstart = 1: start form user-supplied checkpoint files. The working directory must contain molcas_canorb_fiedler.txt and CheMPS2_canorb_MPSx.h5 (x=0 for the ground state, 1 for the first excited state, etc). If these files are not in the working directory, a warning is printed at the beginning of the calculation and restart is skipped (start from scratch). DMREstart = 2 (Not recommended): start form previous checkpoint files with natural orbitals. DMREstart = 2 is not recommended since this may produce non-optimal energy because the orbital ordering is not optimized. |
A general comment concerning the input orbitals: The orbitals are ordered by symmetry. Within each symmetry block the order is assumed to be: frozen, inactive, active, external (secondary), and deleted. Note that if the Spdelete option has been used in a preceding SCF calculation, the deleted orbitals will automatically be placed as the last ones in each symmetry block.
For calculations of a molecule in a reaction field see section
of the present manual and section of the examples manual.
8.41.5.2 Input example
The following example shows the input to the
RASSCF program for a calculation on the water molecule. The calculation is
performed in C2v symmetry (symmetries: a1, b2, b1, a2, where the two
last species are antisymmetric with respect to the molecular plane). Inactive
orbitals are 1a1 (oxygen 1s) 2a1 (oxygen 2s) and
1b1 (the 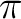 lone-pair orbital). Two bonding and two anti-bonding
OH orbitals are active, a1 and b2 symmetries. The calculation is
performed for the 1A1 ground state. Note that no information about basis set,
geometry, etc has to be given. Such information is supplied by the
SEWARD integral program via the one-electron integral file ONEINT.
lone-pair orbital). Two bonding and two anti-bonding
OH orbitals are active, a1 and b2 symmetries. The calculation is
performed for the 1A1 ground state. Note that no information about basis set,
geometry, etc has to be given. Such information is supplied by the
SEWARD integral program via the one-electron integral file ONEINT.
&RASSCF
Title= Water molecule. Active orbitals OH and OH* in both symmetries
Spin = 1
Symmetry = 1
Inactive = 2 0 1 0
Ras2 = 2 2 0 0
The following input is an example of how to use the RASSCF program to run MC-PDFT calculations:
&RASSCF
Ras2
1 0 0 0 1 0 0 0
>>COPY $CurrDir/$Project.JobIph JOBOLD
&RASSCF
JOBIPH
CIRESTART
CIONLY
Ras2
1 0 0 0 1 0 0 0
KSDFT
ROKS
TPBE
The first RASSCF run is a standard CASSCF calculation that leads to variationally optimized orbitals and CI coefficients.
The second call to the RASSCF input will use the CI vector and the orbitals previously optimized. The second RASSCF will
require the CIONLY keyword as the MC-PDFT is currently not compatible with SCF. KSDFT ROKS and the functional choice will
provide MC-PDFT energies.
More advanced examples can be found in the tutorial section of the manual.
Input example for DMRG-CASSCF with Molcas-CheMPS2 interface:
&RASSCF
Title= Water molecule. Active orbitals OH and OH* in both symmetries
Spin = 1
Symmetry = 1
Inactive = 2 0 1 0
Ras2 = 2 2 0 0
DMRG = 500
3RDM
Next: 8.42 rassi Up: 8. Programs Previous: 8.40 quater

