




















MOLCAS manual: Next: 8.14 expbas Up: 8. Programs Previous: 8.12 embq
|
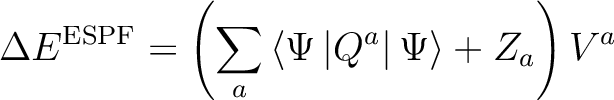 |
(8.1) |
NOTE: Always run SEWARD + ESPF. If not, very strange results may happen due to interactions counted twice or more!
NOTE: Symmetry is ignored since the external potential usually breaks the one given in GATEWAY.
If no external potential is given, the ESPF module can be used to compute atomic point charges fitted to the electrostatic potential produced by the nuclei and electrons.
8.13.2 ESPF and QM/MM
Whereas the ESPF method can be used standalone, it has been developed for hybrid quantum mechanics/molecular mechanics (QM/MM) computations, in which an extended molecular system is divided into two subsystems: the 'active' center described by any QM method and its surroundings qualitatively treated with an empirical atomic forcefield. The current implementation can be used with either a modified version of the TINKER program or with the GROMACS program as MM code.
8.13.2.0.1 Using MOLCAS together with Tinker.
In order to obtain the modified TINKER code, you must run the "molcas get_tinker" command.The current patched version of TINKER8.1 is 6.3.3.
IMPORTANT: The environment variable TINKER must point to the directory in which the TINKER executable binaries are stored (usually in $MOLCAS/tinker/bin).
The most convenient way to define (i) the QM and MM subsystems and (ii) which atoms are to be known by MOLCAS (all the QM ones and some MM ones, see below) requires to simply add the keyword TINKER in GATEWAY. This way, GATEWAY will ask TINKER to pass it all information needed.
Alternatively, an old input style can be used in GATEWAY, using the following syntax:
Basis set
X..... / MM
name x y z Angstrom
Charge
q
End of basis
where q gives its MM point charge value, name is its name and x, y, z are its coordinates given in au or in Å if the ANGSTROM keyword is given. This way, a MM atom is simply a QM atom without basis functions and with a non-integer atomic charge. Usually only MM atoms needed to define some constrained link atoms (see below) positions are included, however this is not mandatory.
8.13.2.0.2 Using MOLCAS together with Gromacs.
The interface to GROMACS differs from the TINKER interface in that the MM code is not run as a separate program but included in MOLCAS as a library. In this way, the communication between the QM and MM codes is handled by simple function calls instead of using data files. The interface is automatically installed along with MOLCAS provided that the GROMACS library (currently a development version8.2) is available at configuration time8.3. Instructions how to install the GROMACS library can be found at the official web site8.4. Make sure that the installation is done in double precision since this is the precision used by MOLCAS. Also make sure to source the GROMACS GMXR script in your shell startup file, otherwise the MOLCAS configuration procedure will not be able to detect the relevant library path.
The recommended (and the only verified) approach of using the MOLCAS/GROMACS interface is to define the full QM+MM system in the GROMACS input. The system definition can then be imported into MOLCAS by adding the keyword GROMACS in GATEWAY (see section ![[*]](crossref.png) for details). For efficiency reasons, the MOLCAS part of the interface separates the MM subsystem into two different atom types: inner MM atoms and outer MM atoms. These are completely equivalent as far as interactions are concerned. However, whereas the coordinates of the inner MM atoms are stored and updated using MOLCAS standard mechanism, the outer MM atoms are handled using a mechanism specifically designed with large systems in mind. The division into inner and outer MM atoms can be controlled with options to the GROMACS keyword in GATEWAY (see section ).
for details). For efficiency reasons, the MOLCAS part of the interface separates the MM subsystem into two different atom types: inner MM atoms and outer MM atoms. These are completely equivalent as far as interactions are concerned. However, whereas the coordinates of the inner MM atoms are stored and updated using MOLCAS standard mechanism, the outer MM atoms are handled using a mechanism specifically designed with large systems in mind. The division into inner and outer MM atoms can be controlled with options to the GROMACS keyword in GATEWAY (see section ).
Please note that the MOLCAS/GROMACS interface is still under development and is currently provided for evaluation purposes only.
8.13.2.0.3 The QM/MM method.
The Hamiltonian of the full QM/MM system is divided into three terms:| H=HQM+HMM+HQM/MM | (8.2) |
The first one describes the QM part as it would be in vacuo, the second one describes the surroundings using a classical MM forcefield and the last one deals with the interactions between the QM and the MM subsystems. In its usual formulation, the last term is (for q point charges interacting with N nuclei and n electrons):
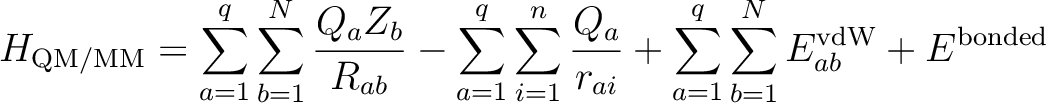 |
(8.3) |
The usual forcefields use the "1-4 condition" to separate the bonded interactions (stretching, bending, torsion) from the non-bonded ones (electrostatic and vdw). This means that the non-bonded potentials are applied only if atoms are separated by 3 bonds or more. As for the QM/MM interactions, this procedure is kept with the exception that all the QM atoms experience the electrostatic potential generated by all the MM point charges (the QM/MM frontier case is considered later).
NOTE: Starting with MOLCAS 8, all MM point charges interact with the QM charge distribution using the ESPF method (at variance with previous MOLCAS versions in which the few MM atoms defined in GATEWAY were interacting directly with the QM electrons and nuclei).
8.13.2.0.4 Link atoms.
When no bonds are involved between the QM and the MM parts, the QM/MM frontier definition is obvious and only the electrostatic and vdw interactions are taken into account. However, if one or several chemical bonds exist, the definition of a smooth but realistic frontier is needed. Several schemes, more or less sophisticated, have been proposed. In the current implementation, only the most basic one, the link atom (LA) approach is included. In the LA approach, each QM/MM bond that should be cut is saturated with a monovalent atom — most often a hydrogen atom — on the QM side. The position of a link atom is often restrained: frozen distance from the corresponding QM frontier atom and always on the segment defined by the two frontier atoms (Morokuma's method, selected by the LAMOROKUMA keyword).
From the macromolecular point of view, link atoms do not exist, i.e. they should not interact with the MM part. However, this leads to severe overpolarization of the frontier, due to unbalanced interactions. Hence interactions between the link atoms and the MM potential is kept. To remove problems that may arise from too strong interactions between a link atom and the closest MM point charges, these point charges may be spread in the MM neighborhood. For instance, in a protein, this procedure is mainly justified if the MM frontier atom is an  carbon (Amber or Charmm-type forcefields usually set these point charges close to zero).
carbon (Amber or Charmm-type forcefields usually set these point charges close to zero).
8.13.2.0.5 Geometry optimization – microiterations.
In a QM/MM geometry optimization job, a MOLCAS step costs as hundreds of TINKER or GROMACS steps. Thus it is very convenient to use the microiteration technique, that is, converging the MM subsystem geometry every MOLCAS step. In the case of TINKER, this is requested in the TINKER keyword file, whereas if GROMACS is used, it is requested directly in ESPF. In order to improve the optimization convergence, an improved QM/MM Hessian can be built in SLAPAF using the RHIDDEN keyword (note that adding the keyword CARTESIAN may help too).
8.13.3 Dependencies
The ESPF program depends on SEWARD for modifying the core Hamiltonian matrix and on ALASKA for computing the extra contributions to the gradient.
8.13.4 Files
ESPF will use the following input
files: RYSRW, ABDATA, RUNFILE, ONEINT (for more information see ).
In addition, ESPF uses ESPFINP (the ESPF input file) and SEWARINP (the Seward input file).
Please note that the external potential can be given within a file, separated from the ESPF input file.
In calculations using the MOLCAS/GROMACS interface, ESPF will additionally need access to the GROMACS tpr file.
8.13.4.1 Intermediate files
All the intermediate files are related to the use of ESPF together TINKER. The files allow for communication between the ESPF program and the MM code. MOLCAS uses one file to pass the QM atoms coordinates and ESPF-derived point charges to TINKER. TINKER uses the same file to pass the external potential, the MM-only energy and gradient components to MOLCAS.
| File | Contents |
| TINKER.LOG | The log file of the Tinker run. |
| $Project.xyz | The coordinate file for TINKER. |
| $Project.key | The keyword file for TINKER. |
| $Project.qmmm | The communication file between MOLCAS and TINKER. |
8.13.4.2 Output files
| File | Contents |
| ONEINT | One-electron integral file generated by the SEWARD program. |
| RUNFILE | Communication file for subsequent programs. |
| ESPF.DATA | Ascii file containing some specific informations needed for subsequent calls to the ESPF module. |
| GMX.LOG | Logfile for the GROMACS library routines. |
8.13.5 Input
Below follows a description of the input to ESPF.
In addition to the keywords and the comment lines the input may contain blank lines. The input for each module is preceded by its name like:
&ESPF
Compulsory keywords
| Keyword | Meaning |
| EXTErnal | Specify how the external potential is given. This keyword is compulsory in the first run of ESPF. On the next line, one integer or a text string must be given:
|
Optional keywords
| Keyword | Meaning |
| TITLE | Title of the job. |
| MULTipoleorder | Multipolar order of the ESPF operators. For TINKER, allowed values are 0 (charge-like) or 1 (charge- and dipole-like). For GROMACS, only 0 is allowed. Default value is 0. |
| GRID | Modify the grid specifications. The grid is made of points belonging to molecular surfaces defined according to the van der Waals radii of each quantum atom. Two schemes are available. The first one is the GEPOL procedure, as implemented into the PCM SCRF method. The other one is called PNT and is the default. On the next line, first select the method with the GEPOL or PNT option. On the same line, one integer number and one real number are given if PNT is selected. The first one gives the maximum number of shells around the van der Waals surface of the quantum atoms. The second one gives the distance between the shells. Note that all points within the van der Waals envelope are discarded to avoid the penetration effects. Default values are 4 shells separated by 1 Å. Alternatively, if GEPOL is selected, the same line must contain 1 integer indicating the number of surfaces to be computed (must be < 6). |
| SHOW | Requires the printing of the ESPF.DATA file. |
| LAMOrokuma | Activate the Morokuma scheme for scaling the link atom positions in a QM/MM calculation. Note that in the case of TINKER, the scaling factor is currently hard-coded and is determined from the radii of the atoms involved in the QM/MM frontier bond. This differs from the GROMACS interface in which this factor must be provided by the user through the LINKATOMS keyword in GATEWAY. |
| MMITerations | Maximum number of microiterations used to optimize the outer MM atoms in a MOLCAS/GROMACS run. The default is 0, which disables microiterations and leaves the outer MM atoms frozen. For the TINKER interface, microiterations are requested in the TINKER keyword file. |
| MMCOnvergence | Convergence threshold for the MM microiterations (GROMACS only). The optimization of the (outer) MM atoms will stop when the maximum force component is smaller than this number, in atomic units. The default is 0.001 atomic units (50 kJ/mol/nm). |
8.13.6 Examples
8.13.6.1 ESPF example
This is a typical input for the calculation of the energy and the gradient of a glycine molecule feeling the external potential of 209 TIP3P water molecules.
&Gateway
Basis set
C.sto-3g.....
C1 1.11820 0.72542 -2.75821 Angstrom
C2 1.20948 0.66728 -1.25125 Angstrom
End of basis
Basis set
O.sto-3g.....
O1 2.19794 1.10343 -0.67629 Angstrom
End of basis
Basis set
H.sto-3g.....
H1 2.02325 1.18861 -3.14886 Angstrom
H2 0.25129 1.31794 -3.04374 Angstrom
H3 1.02458 -0.28460 -3.15222 Angstrom
End of basis
Basis set
N.sto-3g.....
N1 0.17609 0.12714 -0.61129 Angstrom
End of basis
Basis set
C.sto-3g.....
C3 0.09389 -0.01123 0.84259 Angstrom
C4 -1.21244 -0.67109 1.28727 Angstrom
End of basis
Basis set
O.sto-3g.....
O2 -2.06502 -1.02710 0.48964 Angstrom
End of basis
Basis set
H.sto-3g.....
H4 -0.61006 -0.21446 -1.14521 Angstrom
H5 0.92981 -0.61562 1.19497 Angstrom
H6 0.16338 0.97444 1.30285 Angstrom
End of basis
Basis set
N.sto-3g.....
N2 -1.41884 -0.85884 2.57374 Angstrom
End of basis
Basis set
H.sto-3g.....
H7 -0.73630 -0.57661 3.25250 Angstrom
H8 -2.28943 -1.29548 2.82140 Angstrom
End of basis
&seward
&espf
MultipoleOrder = 0
External = 0
1 -0.048 -0.002 -0.006 -0.001 0.007 -0.009 0.002 -0.001 0.001 -0.001
2 -0.047 -0.002 0.001 -0.002 0.003 0.000 -0.004 0.000 -0.001 0.000
3 -0.053 0.004 0.000 -0.011 0.002 0.002 -0.004 0.002 0.003 -0.007
4 -0.046 0.011 -0.009 -0.001 0.006 -0.005 -0.001 0.003 0.003 -0.004
5 -0.042 -0.016 -0.011 -0.006 0.005 -0.007 0.003 -0.004 -0.001 -0.005
6 -0.050 0.000 0.008 0.001 0.006 -0.006 0.000 -0.002 0.000 -0.001
7 -0.039 -0.008 0.001 0.000 0.001 -0.002 0.001 -0.001 -0.001 -0.001
8 -0.032 -0.007 -0.002 0.004 0.002 -0.003 0.001 -0.002 0.002 -0.001
9 -0.011 -0.009 0.004 0.001 0.002 0.000 -0.002 -0.001 0.001 0.001
10 0.000 -0.011 0.003 0.004 0.001 0.002 -0.003 0.001 -0.001 0.001
11 -0.028 -0.008 0.004 -0.001 -0.001 -0.002 0.002 -0.001 0.001 -0.002
12 -0.026 0.003 -0.008 0.014 0.002 -0.001 -0.001 -0.008 0.006 -0.009
13 -0.037 -0.008 -0.003 0.004 -0.007 0.007 0.000 0.001 0.007 -0.001
14 -0.016 -0.007 0.007 -0.008 0.003 0.003 -0.006 0.000 0.002 0.002
15 -0.025 0.003 0.012 -0.007 0.003 -0.001 -0.002 -0.006 0.005 0.009
16 -0.010 -0.011 0.000 -0.014 0.001 0.007 -0.008 0.001 0.000 -0.001
&scf
Charge = 0
&alaska
8.13.6.2 MOLCAS/Tinker example
A typical start for a QM/MM calculation with the MOLCAS/TINKER interface is given in the following input. It is quite general since all the information related to the QM and MM subsystem definitions are already included in the TINKER key file.
> EXPORT TINKER=$MOLCAS/tinker/bin_qmmm
> COPY $PATH_TO/$Project.xyz $WorkDir/$Project.xyz
> COPY $PATH_TO/$Project.key $WorkDir/$Project.key
&Gateway
Tinker
Basis = STO-3G
Group = Nosym
&Seward
&Espf
External = Tinker
LAMorok
This can be used, e.g. with the following TINKER files. In this example, the asparate anion is cut into two pieces, the QM subsystem contains the end of the side-chain until the  carbon atom. There is a link atom between the QM and MM carbon atoms.
carbon atom. There is a link atom between the QM and MM carbon atoms.
QMMM.xyz
16 ASP 1 N3 -0.040452 0.189961 0.173219 448 2 6 14 15 2 CT -0.011045 -0.060807 1.622395 449 1 3 7 11 3 C 1.446535 -0.110535 2.028518 450 2 4 5 4 O 1.902105 0.960982 2.409042 452 3 5 O 2.137861 -0.898168 1.387158 452 3 6 H 0.559257 -0.496270 -0.262338 451 1 7 CT -0.789906 -1.336520 1.982558 216 2 8 12 13 8 C -2.256402 -1.184505 1.571038 218 7 9 10 9 O2 -2.460769 -0.949098 0.356151 219 8 10 O2 -3.120135 -1.188969 2.465678 219 8 11 H1 -0.478878 0.773493 2.145163 453 2 12 HC -0.356094 -2.194944 1.466324 217 7 13 HC -0.720511 -1.505463 3.058628 217 7 14 H -0.996208 0.061130 -0.151911 451 1 15 H 0.304306 1.116522 -0.018698 451 1 16 HLA -0.283317 -0.506767 1.748300 2999 2 7
QMMM.key
* Change $PATH_TO_TINKER parameters $PATH_TO_TINKER/params/amber99.prm QMMM 8 QM -8 10 7 12 13 MM 2 LA 16 * Add the atom type for the LA atom 2999 99 HLA "Hydrogen Link Atom" 1 1.008 0 charge -2 0.0 charge -11 0.0 QMMM-MICROITERATION ON
8.13.6.3 MOLCAS/Gromacs example
To be provided soon.Next: 8.14 expbas Up: 8. Programs Previous: 8.12 embq

