




















MOLCAS manual: Next: 10.8 Extra information about basis Up: 10. Examples Previous: 10.6 Solvent models.
MOLCAS-8.1 is intended for calculations on systems including all atoms of the
periodic table. This is only possible if relativistic effects can be added in a
way that is accurate and at the same time applies to all the methods used in
MOLCAS-8.1, in particular the CASSCF and CASPT2 approaches. MOLCAS-8.1
includes relativistic effects within the same wave function framework as used in
non-relativistic calculations. This has been possible by partitioning the
relativistic effects into two parts: the scalar relativistic effects and
spin-orbit coupling. This partitioning is based on the Douglas-Kroll (DK)
transformation of the relativistic Hamiltonian [286,287].
|
| Spin | C2 sym | Labels in linear symmetry | No. of states |
| 2 | 1 | 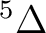 , ,
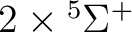 , , 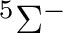 |
5 |
| 2 | 2 |
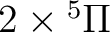 |
4 |
| 1 | 1 |
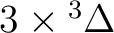 , ,
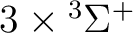 , ,
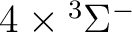 |
13 |
| 1 | 2 |
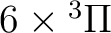 , , 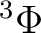 |
14 |
| 0 | 1 | 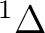 , ,
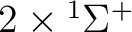 , , 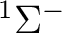 |
5 |
| 0 | 2 |
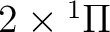 |
4 |
The total number of states is 45. One thus has to perform 6 CASSCF (and
MS-CASPT2) calculations according to the spin and symmetries given in the table.
The RASSI-SO calculation will yield 134 levels with 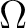 ranging from 0 to
4. Only the lower of these levels will be accurate because of the limitations in
the selection of electronic states.
ranging from 0 to
4. Only the lower of these levels will be accurate because of the limitations in
the selection of electronic states.
The active space used in these calculations is 6s,6p for Pb and 2p for O. This is the natural choice and works well for all main group elements in most molecules. The s-orbital should be active in groups IIa-Va, but may be left inactive for the heavier atoms (groups VIa-VIIa). The ANO-RCC basis sets have been constructed to include correlation of the semi-core electrons. For Pb they are the 5d, which should then not be frozen in the CASPT2 calculations. All other core electrons should be frozen, because there are no basis functions to describe their correlation. Including them in the correlation treatment may lead to large BSSE errors.
The input file for these calculations is quite lengthy, so we show here only one
set of CASSCF/CASPT2 calculations but the whole RASSI input
for all six cases.
&GATEWAY
Title= PbO
Coord= $CurrDir/PbO.xyz
Basis set
ANO-RCC-VQZP
Group= XY
AngMom
0.00 0.00 0.00
End of Input
&SEWARD
End of Input
&SCF
Title
PbO
Occupied
24 21
Iterations
20
Prorbitals
2 1.d+10
End of Input
&RASSCF
Title
PbO
Symmetry
1
Spin
5
CIROOT
5 5 1
nActEl
8 0 0
Inactive
23 18
Ras2
3 4
Lumorb
THRS
1.0e-8 1.0e-04 1.0e-04
Levshft
1.50
ITERation
200 50
CIMX
200
SDAV
500
End of Input
&CASPT2
Title
PbO
MAXITER
25
FROZEN
19 16
Focktype
G1
Multistate
5 1 2 3 4 5
Imaginary Shift
0.1
End of Input
>> COPY $Project.JobMix $CurrDir/JobMix.12
&RASSCF
Title
PbO
Symmetry
2
Spin
5
CIROOT
4 4 1
nActEl
8 0 0
Inactive
23 18
Ras2
3 4
Lumorb
THRS
1.0e-8 1.0e-04 1.0e-04
Levshft
1.50
ITERation
200 50
CIMX
200
SDAV
500
End of Input
&CASPT2
Title
PbO
MAXITER
25
FROZEN
19 16
Focktype
G1
Multistate
4 1 2 3 4
Imaginary Shift
0.1
>> COPY $Project.JobMix $CurrDir/JobMix.22
&RASSCF
Title
PbO
Symmetry
1
Spin
3
CIROOT
13 13 1
nActEl
8 0 0
Inactive
23 18
Ras2
3 4
Lumorb
THRS
1.0e-8 1.0e-04 1.0e-04
Levshft
1.50
ITERation
200 50
CIMX
200
SDAV
500
End of Input
&CASPT2
Title
PbO
MAXITER
25
FROZEN
19 16
Focktype
G1
Multistate
13 1 2 3 4 5 6 7 8 9 10 11 12 13
Imaginary Shift
0.1
End of Input
>> COPY $Project.JobMix $CurrDir/JobMix.11
&RASSCF
Title
PbO
Symmetry
2
Spin
3
CIROOT
14 14 1
nActEl
8 0 0
Inactive
23 18
Ras2
3 4
Lumorb
THRS
1.0e-8 1.0e-04 1.0e-04
Levshft
1.50
ITERation
200 50
CIMX
200
SDAV
500
End of Input
&CASPT2
Title
PbO
MAXITER
25
FROZEN
19 16
Focktype
G1
Multistate
14 1 2 3 4 5 6 7 8 9 10 11 12 13 14
Imaginary Shift
0.1
>> COPY $Project.JobMix $CurrDir/JobMix.21
&RASSCF
Title
PbO
Symmetry
1
Spin
1
CIROOT
5 5 1
nActEl
8 0 0
Inactive
23 18
Ras2
3 4
Lumorb
THRS
1.0e-8 1.0e-04 1.0e-04
Levshft
1.50
ITERation
200 50
CIMX
200
SDAV
500
End of Input
&CASPT2
Title
PbO
MAXITER
25
FROZEN
19 16
Focktype
G1
Multistate
5 1 2 3 4 5
Imaginary Shift
0.1
End of Input
>> COPY $Project.JobMix $CurrDir/JobMix.10
&RASSCF
Title
PbO
Symmetry
2
Spin
1
CIROOT
4 4 1
nActEl
8 0 0
Inactive
23 18
Ras2
3 4
Lumorb
THRS
1.0e-8 1.0e-04 1.0e-04
Levshft
1.50
ITERation
200 50
CIMX
200
SDAV
500
End of Input
&CASPT2
Title
PbO
MAXITER
25
FROZEN
19 16
Focktype
G1
Multistate
4 1 2 3 4
Imaginary Shift
0.1
End of Input
>> COPY $Project.JobMix $CurrDir/JobMix.20
>> COPY $CurrDir/JobMix.12 JOB001
>> COPY $CurrDir/JobMix.11 JOB002
>> COPY $CurrDir/JobMix.21 JOB003
>> COPY $CurrDir/JobMix.10 JOB004
>> COPY $CurrDir/JobMix.22 JOB005
>> COPY $CurrDir/JobMix.20 JOB006
&RASSI
Nrof JobIphs
6 5 13 14 5 4 4
1 2 3 4 5
1 2 3 4 5 6 7 8 9 10 11 12 13
1 2 3 4 5 6 7 8 9 10 11 12 13 14
1 2 3 4 5
1 2 3 4
1 2 3 4
Spin Orbit
Ejob
End of Input
In the above definitions of the JobMix files the labels correspond to
symmetry and spin. Thus JobMix.12 is for quintets (S=2) in symmetry 1,
etc. The keyword Ejob ensures that the MS-CASPT2 energies
from the JobMix files are used as the diagonal elements in the SO
Hamiltonian matrix. The output file of one such calculation is quite lengthy (6
CASSCF/MS-CASPT2 calculations and one RASSI). Important
sections of the RASSI output are the spin-free energies (look for the
word "SPIN-FREE" in the listing) and the SOC energies (found by looking for
"COMPLEX"). The complex SO wave functions are also given and can be used to
analyze the wave function. For linear molecules one wants to know the
values of the different solutions. Here the computed transition moments can be
quite helpful (using the selection rules). It is important in a calculation of
many excited states, as the one above, to check for intruder state problems in
the CASPT2 results.
This example includes a large number of states, because the aim was to compute full potential curves. If one is only interested in the properties near equilibrium, one can safely reduce the number of states. For lighter atoms it is often enough to include the spin-free states that are close in energy in the calculation of the SOC. An intersystem crossing can usually be treated by including only the two crossing states. The choice of basis states for the RASSI calculation depends on the strength of the SO interaction and the energy separation between the states.
The above input is for one distance. The shell script loops over distances according to:
Dist='50.0 10.0 8.00 7.00 6.00 5.50 5.00 4.40 4.20 4.00 3.90 3.80 3.75 3.70
3.65 3.60 3.55 3.50 3.40 3.30 3.10'
for R in $Dist
do
cat $CurrDir/template | sed -e "s/Dist/$R/" >$CurrDir/input
rm -rf $WorkDir
mkdir $WorkDir
cd $WorkDir
echo "R=$R" >>$CurrDir/energies
molcas $CurrDir/input >$CurrDir/out_$R
grep "Reference energy" $CurrDir/out_$R >>$CurrDir/energies
grep "Total energy" $CurrDir/out_$R >>$CurrDir/energies
grep "Reference weight" $CurrDir/out_$R >>$CurrDir/energies
done
Thus, the whole potential curves can be run as one job (provided that there are no problems with intruder states, convergence, etc). Notice that the JOBIPH files for one distance are used as input (JOBOLD) for the next distance. The shell script collects all CASSCF and CASPT2 energies and reference weights in the file energies.
We shall not give any detailed account of the results obtained in the calculation of the properties of the PbO molecule. The reader is referred to the original article for details [291]. However it might be of interest to know that the computed dissociation energy (D0) was 5.0 eV without SOC and 4.0 eV with (experiment is 3.83 eV). The properties at equilibrium are much less affected by SOC: the bond distance is increased with 0.003 Å, the frequency is decreased with 11 cm-1. The results have also been used to assign the 10 lowest excited levels.
Next: 10.8 Extra information about basis Up: 10. Examples Previous: 10.6 Solvent models.

