
The lgv program is used to manipulate molecular structures, build new molecules, and visualize orbitals, densities, and related properties. Here, we show how structures can be manipulated.
To open a coordinate file, use the command lgv Water.xyz. To create a new file, use the -n flag: lgv -n Water.xyz. If no filename is specified, lgv will open the first coordinate file it finds in the current directory.
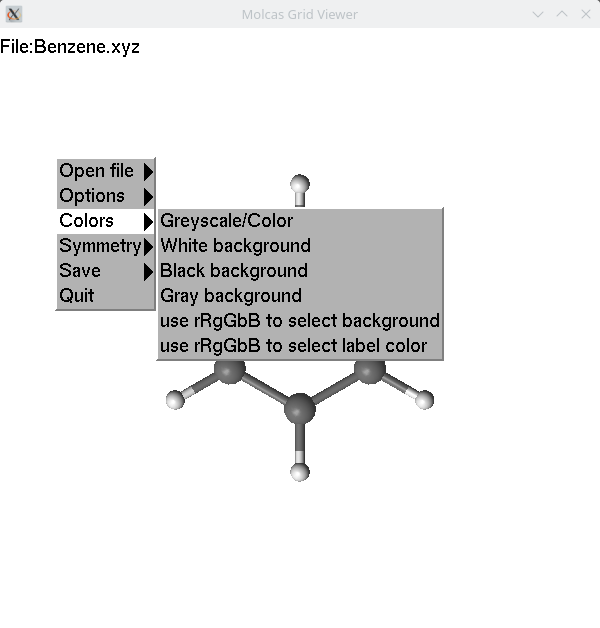
Some common hot keys are shown when the F1 key is pressed. Additional menus are available via right-click.
In order to modify coordinates, we have to select one,
two, three or four atoms. Selection is made by clicking on an atom.
The first selected atom is covered by a blue-colored net; the following
selected atoms are covered by a magenta-colored net. The number of
selected atoms determines the behavior of lgv.
If only one atom is selected, operations apply to this
atom. If two atoms are selected, operations apply to the
bond connecting these atoms. If three atoms are selected,
operations apply to the angle. Finally, four selected atoms
define the dihedral angle. As a general rule, '+/-' changes the
value of the selected object, PageUp/PageDown changes its property, and = (or
F4) allows you to set the value.
To remove the selection, press the Space
bar or click the middle mouse button. Pressing + or - will
modify the value. For example, if a bond is selected and the user presses the '+'
key, the bond length will increase, so the first selected atom will
move away from the second atom.
Example of selected
atom:
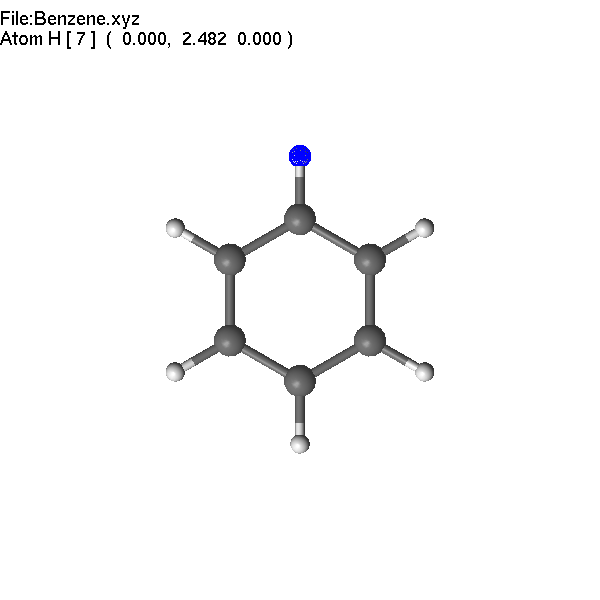
Pressing
F4 (or =) and typing O (note that you do not need to focus into
'edit' window), we change the H atom to O.
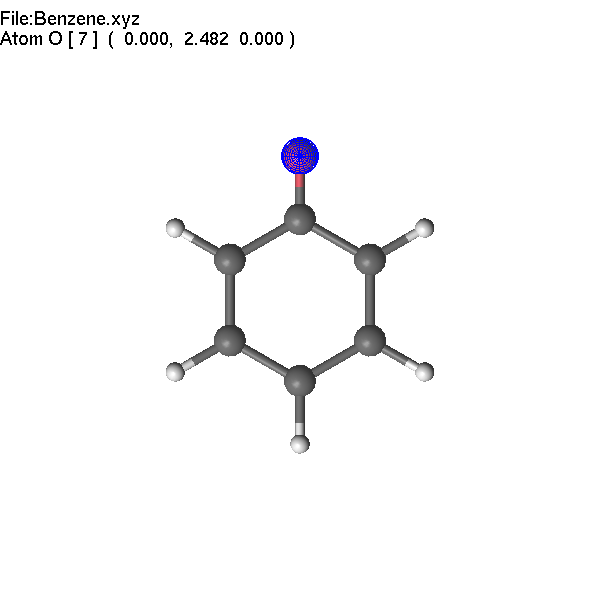
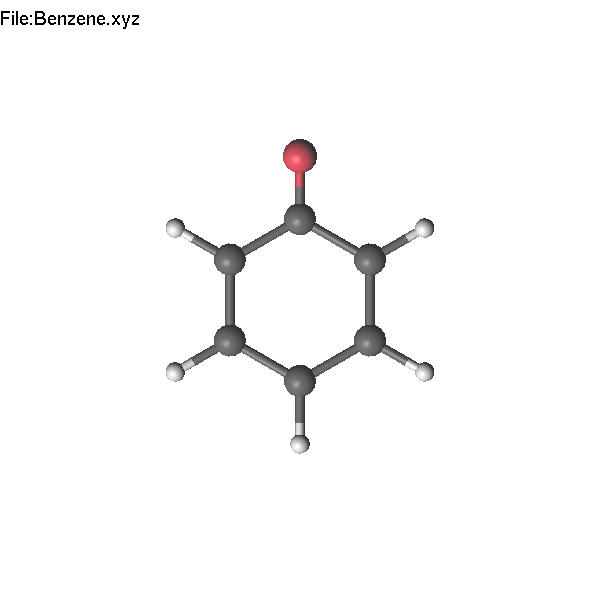
It
is also possible to use PgUp to change the atom name using the list
of the most common elements.
The Backspace key can be used to
undo modifications.
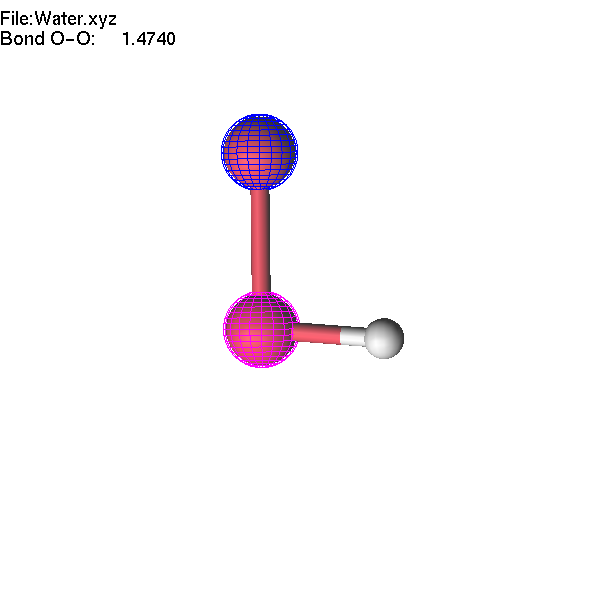
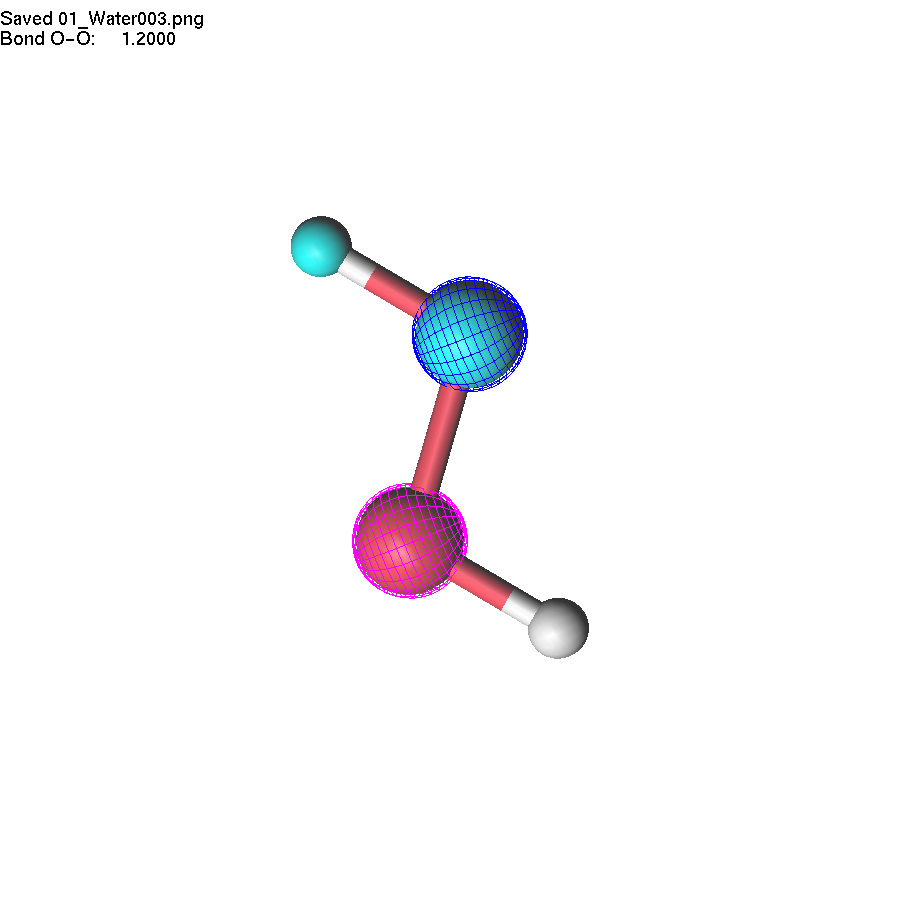
If two atoms are selected, the bond
length can be modified using +/- or =/F4.
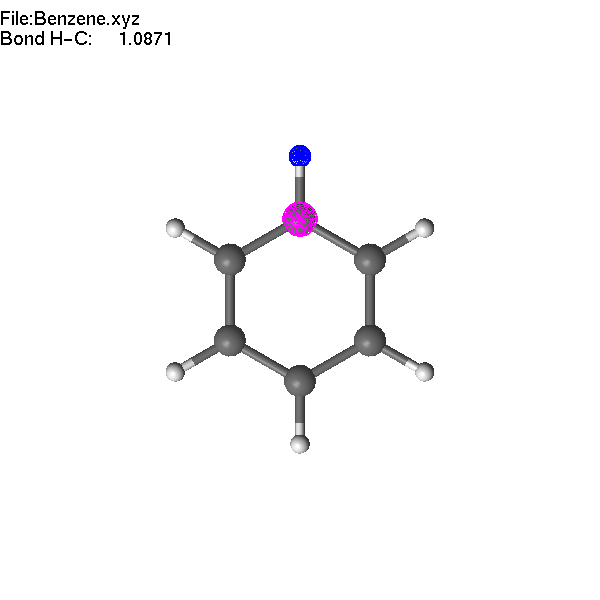
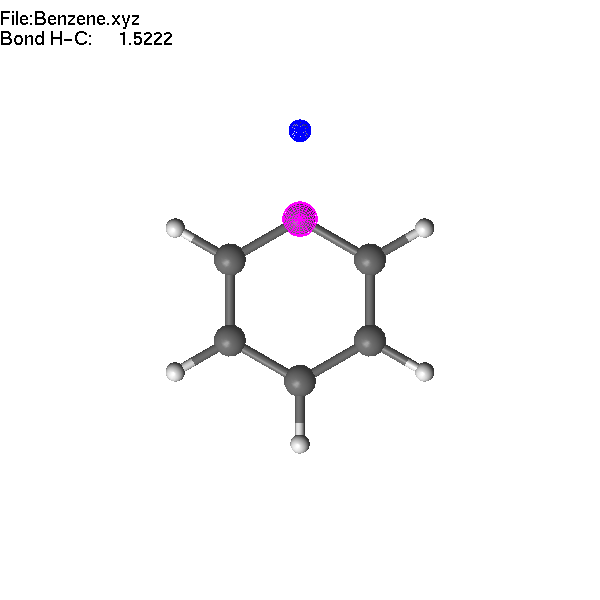
Note
that the first atom (blue) moves during the operation.
Similarly,
if three atoms are selected, the angle value will be modified,
and if four atoms are selected, the dihedral angle will be modified.
Pressing PageUp/PageDown modifies the 'property' of
the selected object. If only one atom is selected - it will change
element name, if a bond is selected, it changes the type of the
bond (single, double, etc.), and if an angle is selected, it changes
the angle to common values.
F2 key can be used to save
the coordinate file. The file name is generated from the
original name by adding a counter. Shift-F2 will overwrite the
original file. These operations work for XYZ, Molden, and lus files.
If one atom is selected, you can use the Delete or Insert key to delete it or insert a new atom. These operations are available for XYZ, Molden, and lus files. Let us make an H2O2 molecule starting from the Water.xyz file.
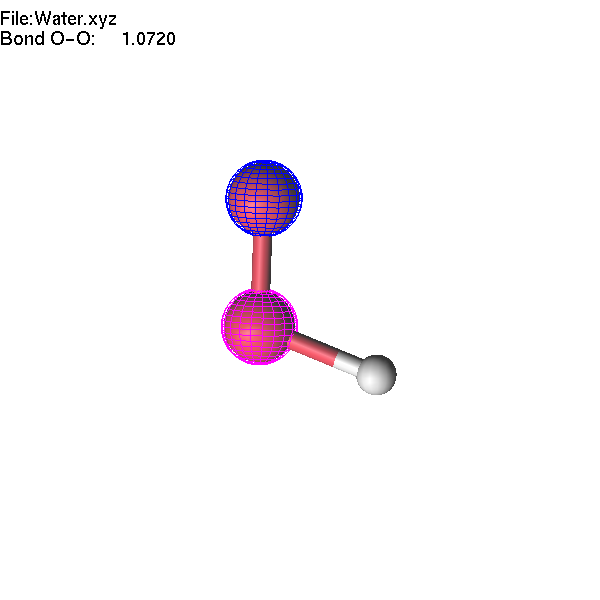
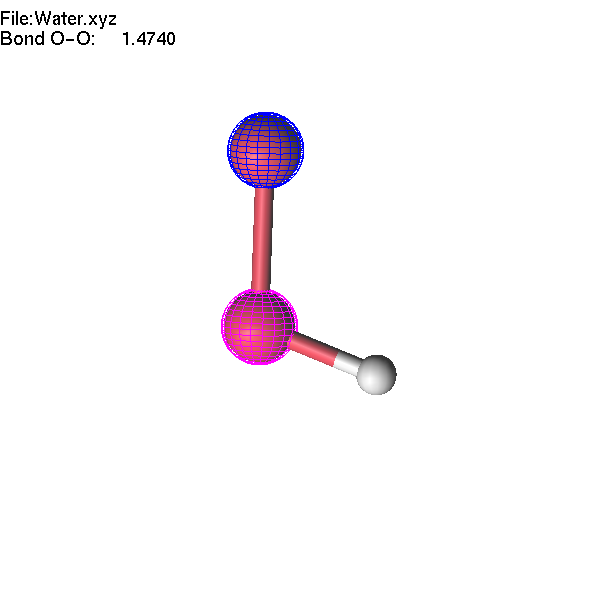
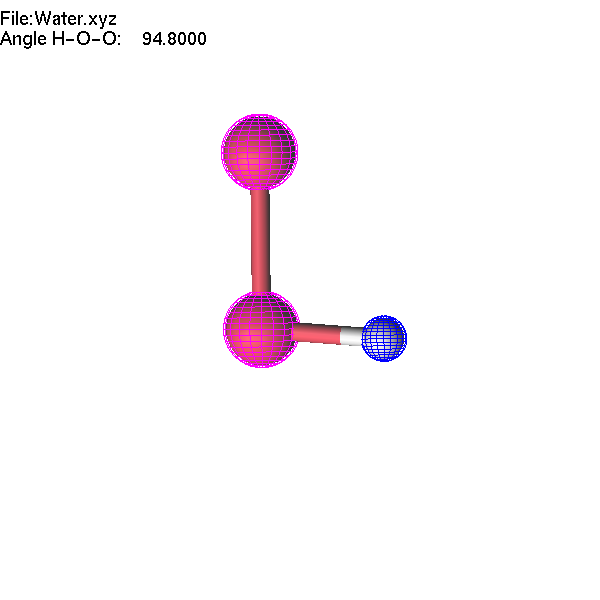
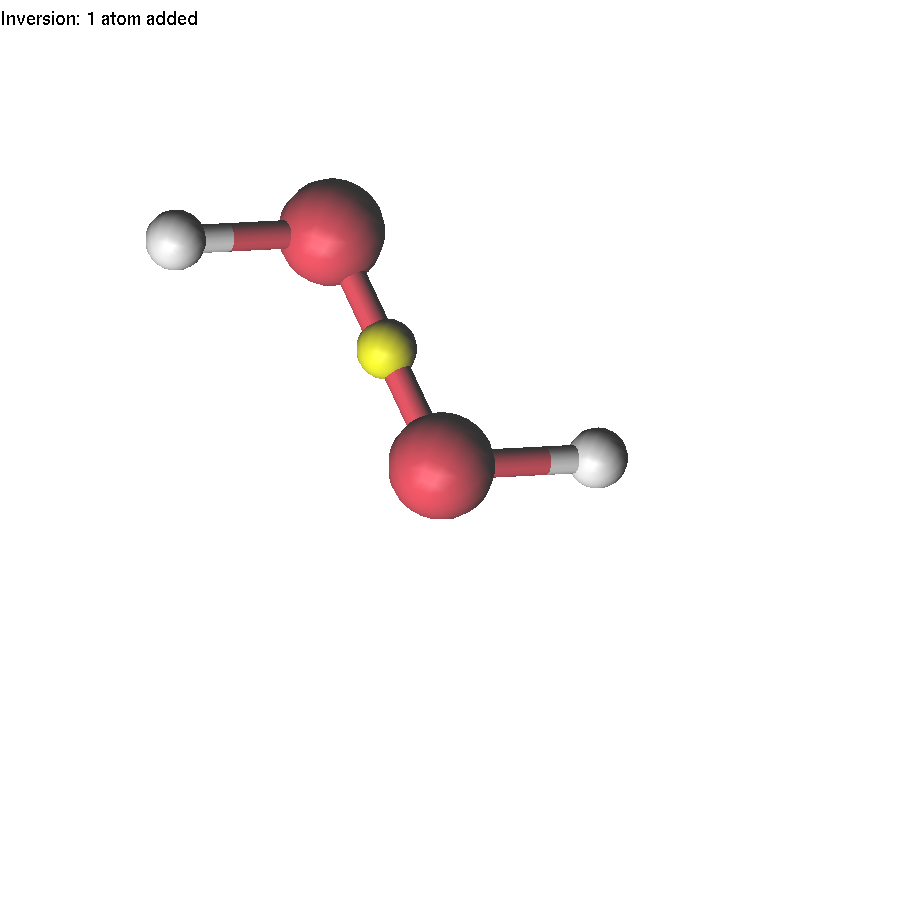
Now we will continue to edit the H2O2 molecule. If we select the O-O bond and change the interatomic distance, only one atom will move. If we want to move a group (O-H), we have to highlight this group first.
There are several ways to highlight (mark) a group of atoms. Probably the simplest way is to use the mouse. Press and hold the Shift key and drag the mouse. All atoms in this rectangular area will be highlighted. Another possibility is to hold the Shift key and click an atom with the mouse; this toggles the status of this atom. It is also possible to use the F7 key to make a selection. If no atom is selected, F7 highlights H atoms. If one atom is selected, F7 highlights its neighbours (atoms connected to this atom), and 'a' highlights all atoms of the same element.
To remove highlighting, press the Space bar or the middle mouse button.
Now, we can make operations with a group of atoms. Select two atoms: first the highlighted oxygen atom, then the non-highlighted one. Then press + until the highlighted fragment is far enough from the other oxygen atom that lgv no longer shows the bond. Note that the decision about which atoms are 'bonded' is based entirely on atomic radii (which can be adjusted).
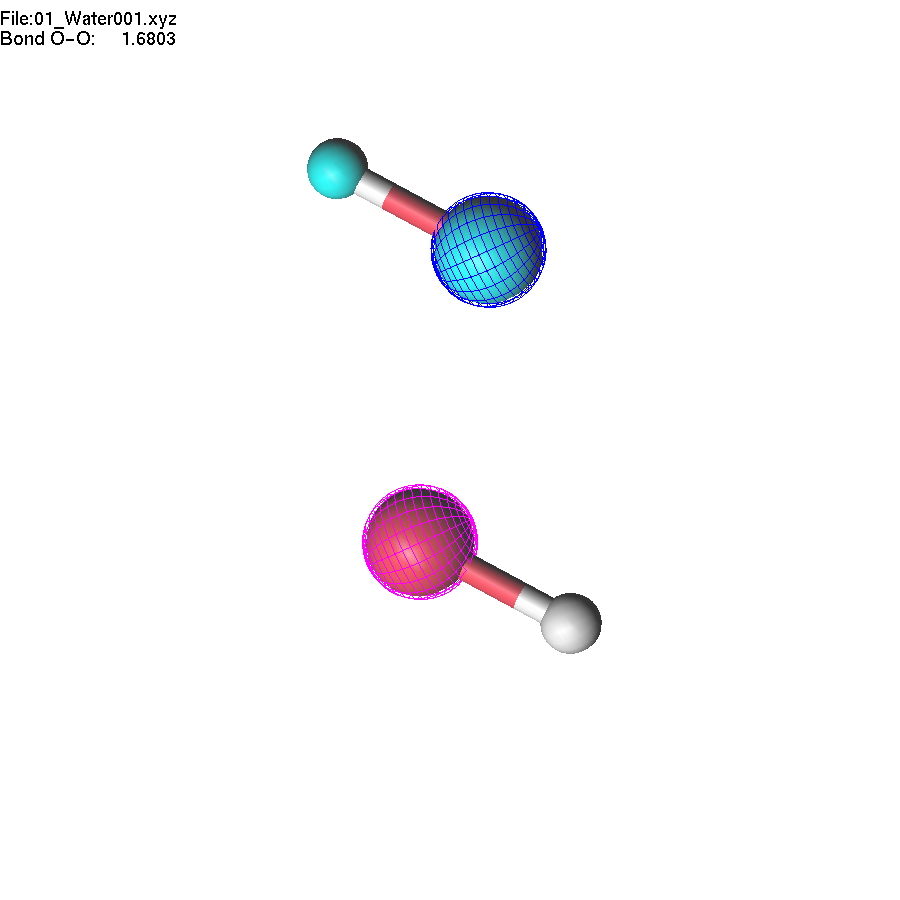
If you change the length of the O-O bond, the whole highlighted group will move accordingly. Note that unselection (Space key, or mouse middle click) will remove selection first, and the second use of the unselect button removes the highlighting of the group.
The modified value (of a bond length or an angle) is shown on the information line of the screen. Sometimes you may want to observe another value while modifying coordinates. To do this, select a bond or an angle and press the F6 key. Now you can make another selection and make modifications in the geometry. In this case, the originally selected value will be watched. Pressing Shift-F6 switches off watch mode.
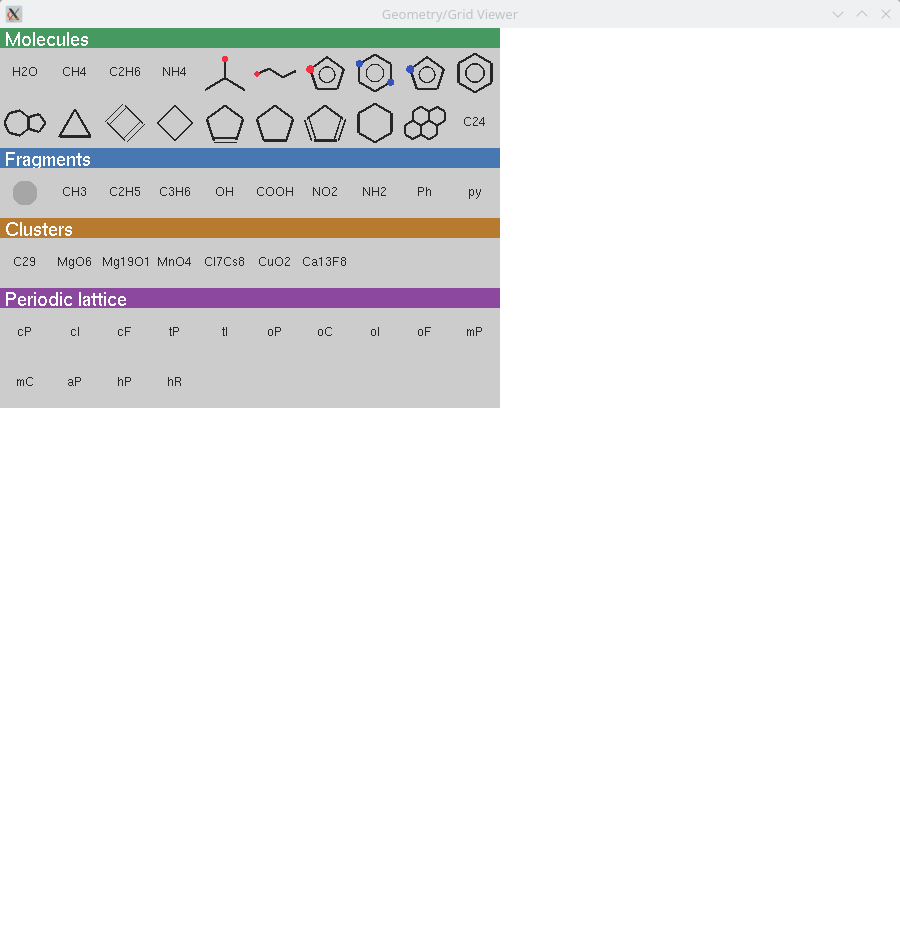
lgv contains a library of molecular fragments that can be added to a molecule. Press F3 to open the fragment picker. Fragments are organised into four visual categories, each with a coloured header bar: Fragments (building blocks with a Q anchor atom for attachment), Molecules (standalone structures inserted near the current molecule), Clusters, and Periodic (structures with lattice vectors). Click an icon to insert the corresponding fragment. For Fragments (with Q anchor): if one atom is selected the fragment is attached near that atom; if no atom is selected it is placed near the current molecule. For Molecules: if one atom is selected the molecule is placed near it; if atoms are present but none selected, click on the scene to choose the placement position; if the scene is empty the molecule is placed at the centre. Once a fragment has been selected, the Insert key re-inserts it without reopening the picker.
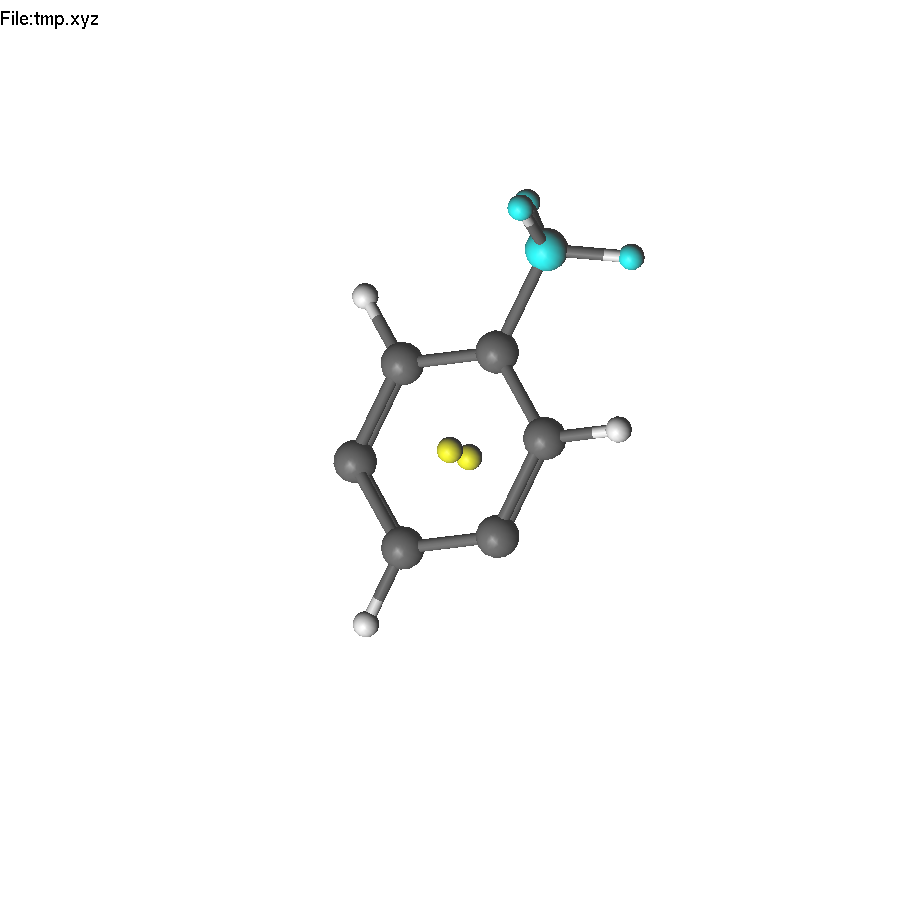
The task: create mesitylene (1,3,5-trimethylbenzene).
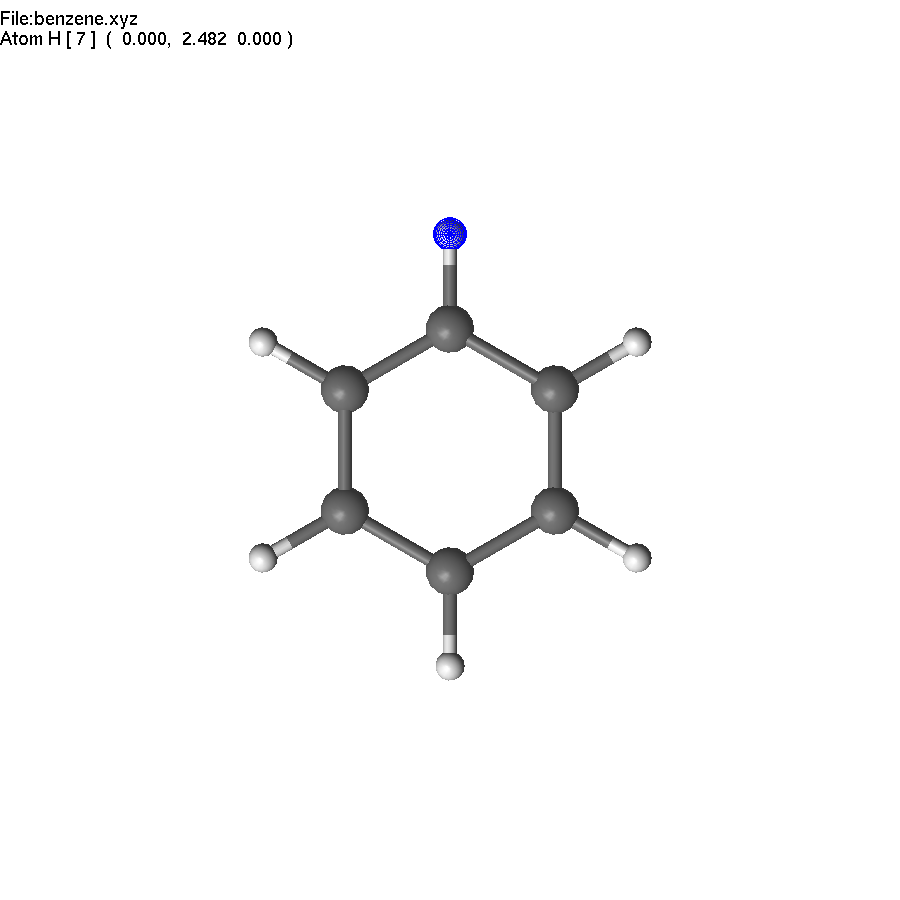
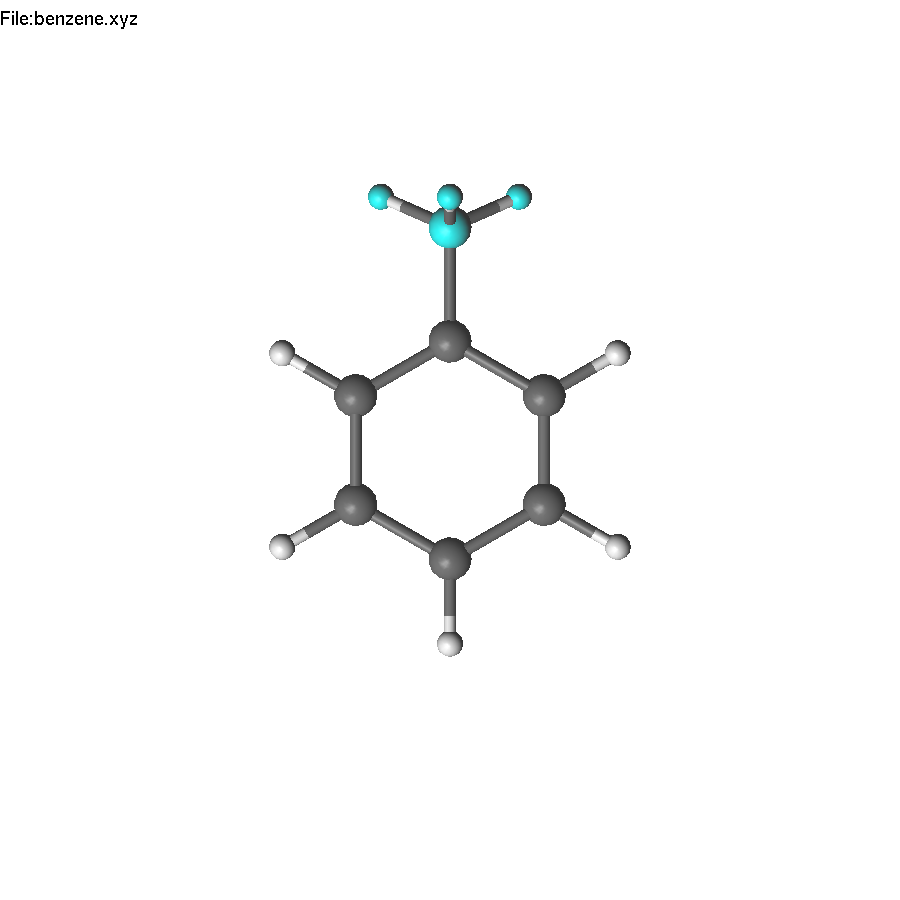
Now we have to repeat the action, or use symmetry! The first step - we need to select a symmetry element, which we can use. It is the principal C6 axis. Unfortunately, there are no atoms on this axis, but we can create some. Select two opposite carbon atoms, and press End. This creates a dummy (yellow) atom in the center of the ring. Unselect (press Space).
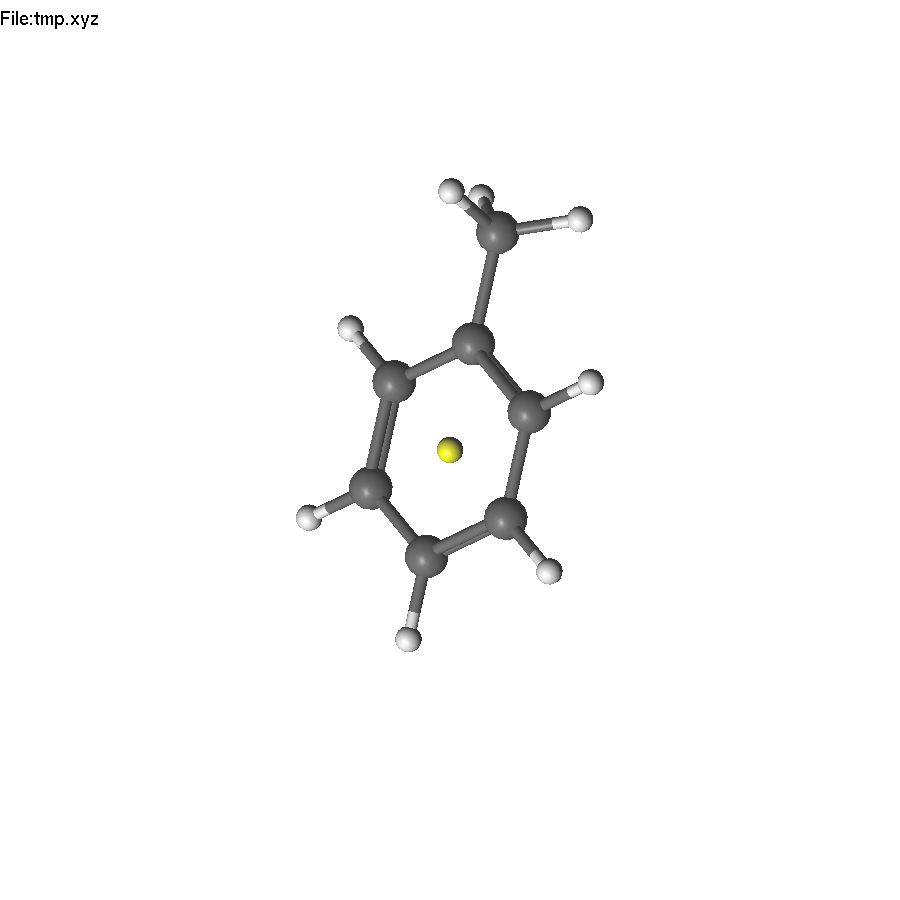
Select the dummy atom and press PageUp, so it will jump from the plane. Repeat the operations to place another dummy atom back to the center.
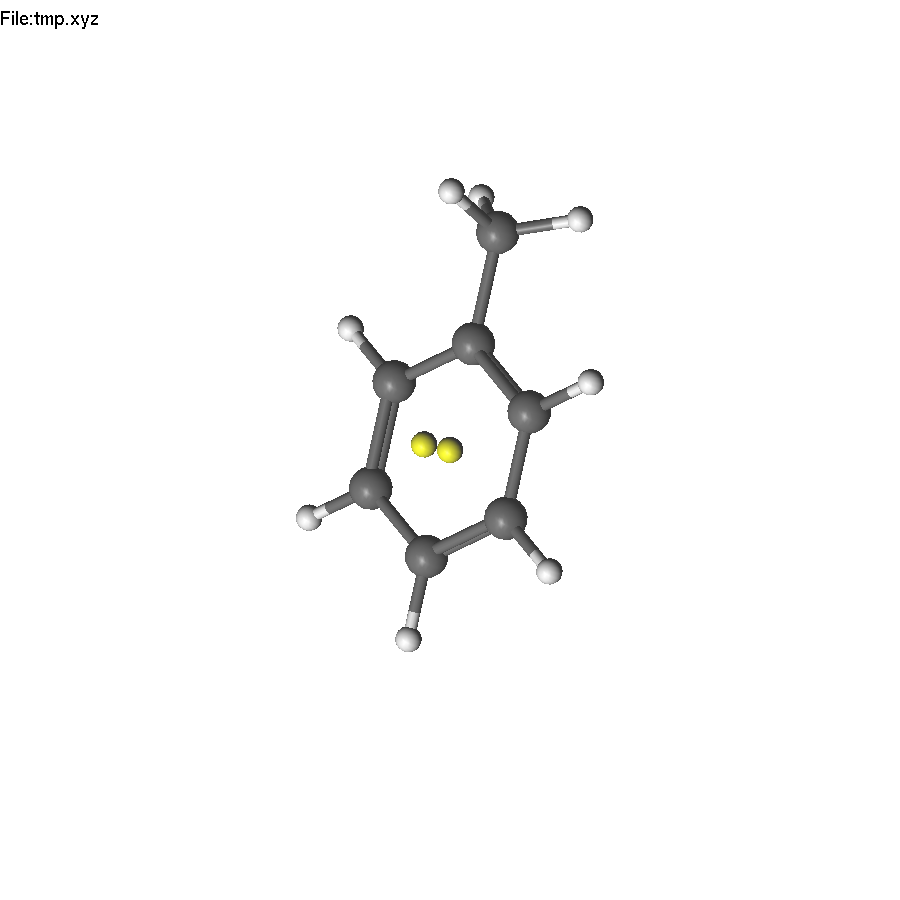
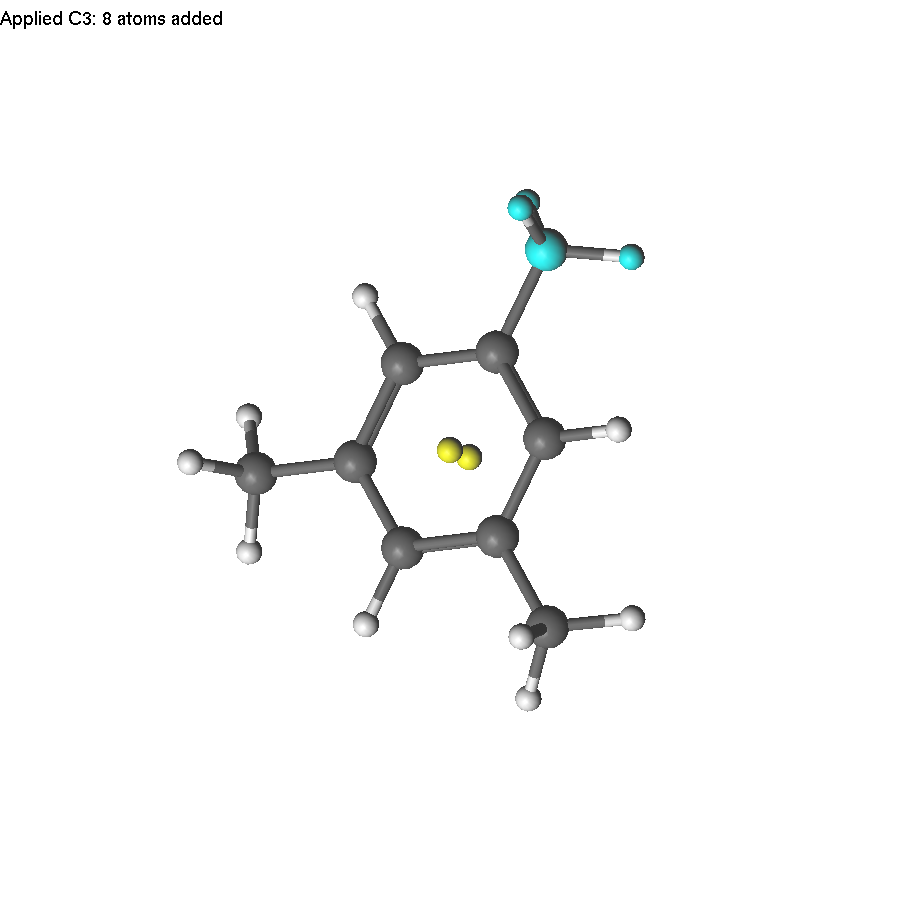
Now select the two yellow atoms and press F8. A help line will appear (since there are several symmetry operations to be applied). Press 3 to use C3. The only problem is that applying the C3 operation also duplicates H atoms, which will become too close to carbon atoms. Therefore, we need to delete them first.
Then select the axis, press F8, and press 3.
To modify coordinates via distances and angles, you may need dummy (reference) atoms. These dummy atoms can be added with the End key. If no atoms are selected, the 'End' key adds dummy atoms located on the X, Y, and Z axes. If a bond is selected, the dummy atom is placed in the middle of the bond. For example, if you have a planar molecule, but it is not oriented according to Cartesian axes, you can add dummy atoms on each axis, highlight all atoms in the molecule, and select a dihedral angle between the plane of the molecule and the desired plane created by the dummy atoms.
The lgv program can recognize the symmetry elements of a molecule, or apply symmetry operations for all, or highlighted atoms in the molecule. If no selection is made, F8 key displays symmetry elements of the molecule. Two symmetry engines are available: msym (default) and findsym. Press Shift-F8 to toggle between them. The fundamental difference between these two codes is that msym detects all symmetry operations, whereas findsym detects only abelian operations.
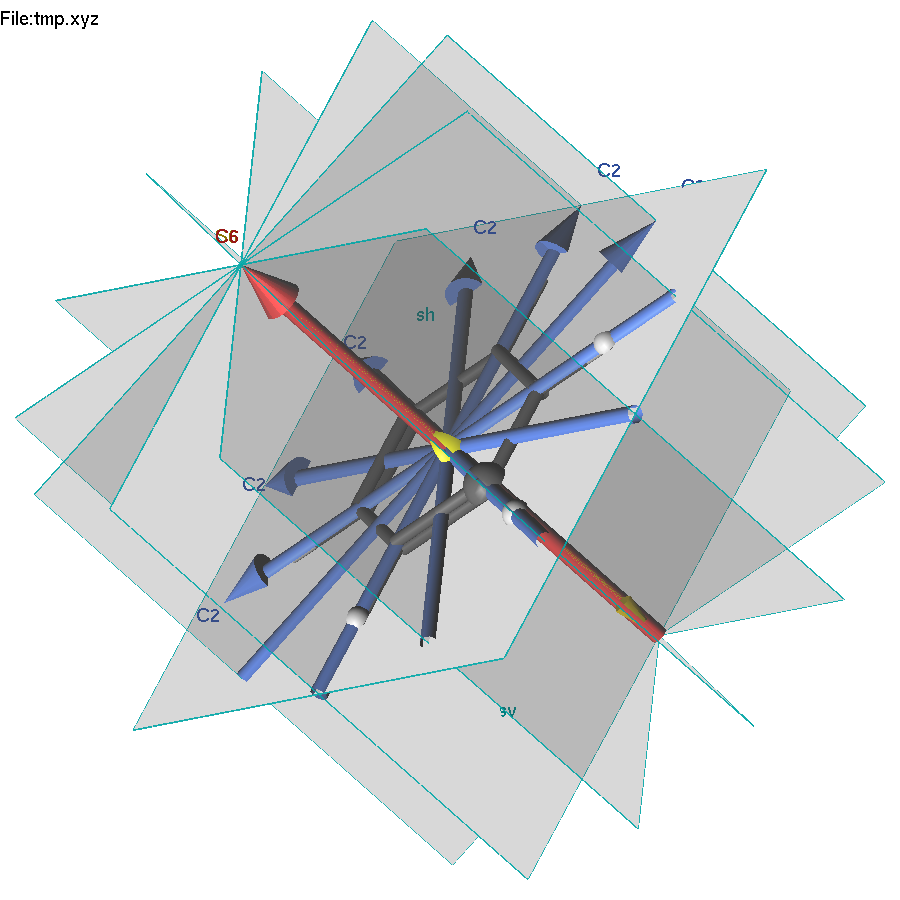
Also
note that if a part of the molecule is highlighted, the symmetry
operation will be applied only to the highlighted part.
In the example
below, we highlighted two atoms and selected two others. F8 translates
the highlighted group by the vector defined by the selected atoms.
lgv can be customized. Press the F9 key to save the current settings. It will create a directory .lgv-config in the current working directory with settings for colors, initial sizes, etc. These files can be edited to change the defaults. To share settings across all working directories, copy .lgv-config/ to $HOME/.lgv-config/. lgv will use the local directory when present, and fall back to the global one otherwise.
lgv can remember a ViewPoint (the current orientation of the molecule) and save it to a file. Later this file can be used to rotate another molecule accordingly. Use keys (v/V) to save or restore Viewpoint.
In some occasions, user would like to change the order of atoms in XYZ file. Although the simplest way to do this includes usage of editor, lgv provides some tools for resorting. If two atoms are selected, and # button pressed, selected atoms will exchange their order. Pressing # reorders atoms.
lgv provides several ways to modify the appearance of atoms. Highlight some atoms (Shift-click or rubber-band), then use the - / + keys to change their colour saturation:
These modifiers are saved with the XYZ file as a suffix on the element name: C-, C--, C--- for pale levels; C+, C++, C+++ for vivid levels. The modifier is preserved across save/load cycles. The A key switches the overall atom rendering style.
Saving coordinates — the Save right-click menu offers three XYZ save variants in addition to the standard F2 / Shift-F2 keys:
lgv automatically detects double and triple bonds from molecular geometry using a valency-based algorithm (ported from LUSCUS):
Bond-order multiplicity explicitly defined in fragment definitions (e.g. C=C in ethylene) takes priority over the auto-detection.
Pressing O (or using the right-click Options menu item Bond order: Auto / Single only) toggles between auto-detection and single-bonds-only mode. The setting is saved with F9.
Double and triple bonds are drawn as two (or three) parallel cylinders. The offset direction can be set in two ways:
Toggle via the right-click Options menu item Bond style: 3D / Billboard. The setting is saved with F9.
After highlighting a group of atoms (e.g. a freshly inserted fragment), pressing E rotates the group around its centroid to minimise the pairwise Coulomb interaction with the rest of the molecule. The centroid position is not changed — only the orientation is optimised.
The algorithm uses a two-pass grid search over SO(3) (ZYZ Euler angles): a coarse pass at 24° steps, followed by a fine pass at 6° steps within ±36° of the coarse minimum. Charges used:
| Atom | Charge | Atom | Charge |
|---|---|---|---|
| H | +1 | O | −2 |
| C | +4 | F, Cl, Br, I | −1 |
| N | +3 | others | 0 |
The feature works for any highlighted group, not only freshly inserted fragments. Press E again to re-run the search from the new orientation.
lgv understands several extensions of the XYZ format. Atom colour modifiers are written as a suffix on the element symbol (see above). At the end of an XYZ file the following #-prefixed lines may appear:
The second line of an XYZ file (the comment line) is displayed as a second status bar line whenever it is non-empty and does not start with produced by. This is useful for files that store a title, formula, or energy in the comment field.
lgv automatically recognises CIF and MOLCAS SEWARD/GATEWAY input files and converts them to XYZ internally — no external tools required.
CIF files (Crystallographic Information Files, extension .cif):
SEWARD/GATEWAY input files (MOLCAS input, typically .inp or any file containing &GATEWAY or &SEWARD):
When a coordinate file contains unit cell vectors (e.g. an XYZ file exported from a periodic calculation, or loaded from a CIF), lgv shows the cell axes on screen and enables cluster generation.
Bond drawing in periodic mode. In mixed-element structures (e.g. CsCl, NaCl) bonds between atoms of the same element are suppressed because they are usually non-physical. In single-element (elemental) structures such as copper or iron, this restriction is lifted and same-element bonds are drawn normally, so that the metal coordination environment is visible.
To generate a cluster of concentric shells around a chosen atom:
Use PageDown to step through smaller clusters (or PageUp for larger ones). The cluster frames are stored in Molden geo format, so all the Molden navigation keys apply.
lgv can drive a MOPAC semi-empirical geometry optimization and reload the result directly, without leaving the viewer. This requires the $MOPAC environment variable to point to the MOPAC binary.
Setup (once, e.g. in ~/.bashrc):
export MOPAC=/opt/mopac/mopac export MOPAC_KEYWORDS="PM7 GNORM=0.01" # optional; default is PM6
Note: this feature is not available in the WebAssembly build.
Watch mode lets you monitor an ongoing calculation in real time. lgv polls the current file and automatically reloads it whenever the file changes on disk.
Watch mode works for XYZ, molden, and multi-frame files. It is particularly useful combined with m (animation) to see the trajectory build up as the calculation runs.
lgv can record a session to a text file and play it back later. This is useful for creating reproducible demonstrations, automated tests, or batch processing scripts.
To start recording, launch lgv with:
lgv --record session.gvr molecule.xyz
All keyboard, mouse, and menu events are written to session.gvr with millisecond timestamps. To play back:
lgv --play session.gvr molecule.xyz
The playback file may also contain script directives (without timestamps) to control the session:
| Directive | Purpose |
| speed <factor> | Playback speed multiplier (e.g. 2.0 = twice as fast). |
| wait <ms> | Insert an extra pause (milliseconds) before the next event. |
| open <filename> | Load a different molecule or grid file during playback. |
| status <text> | Display a message in the lgv status bar. |
| echo <text> | Print a message to standard output. |
Lines beginning with # are treated as comments.
For server-side or automated rendering without a display, lgv can be compiled against the OSMesa off-screen rendering library:
make osmesa (requires libosmesa6-dev)
The OSMesa build renders entirely in memory; no X11 display, GPU, or xvfb is needed. Combined with the -q flag, it exports PNG images and exits:
lgv -q -o all -v molecule.lus
This exports every orbital to separate PNG files named molecule_MO001.png, molecule_MO002.png, etc. The -v flag applies an automatic best viewpoint. The -o flag accepts all, a single number, or a comma-separated list of orbital numbers.
lgv can be compiled to WebAssembly for in-browser visualization:
make -f Makefile.emscripten (requires Emscripten SDK)
The browser build provides the same core visualization features as the native build. Grid density differences (Diff menu in the toolbar) are fully supported: load a second .lus file, choose Minus or Plus and Same-space or Align (Kabsch rigid-body alignment), then click Compute Diff to display the result immediately.
Editing operations are accessible via the toolbar:
The following features are not available in the WebAssembly version:
The JavaScript interface exposes functions such as gv_load_file, gv_compute_diff, gv_key_page_up, gv_save_png, gv_make_clusters, and others, allowing the browser UI to control lgv programmatically. PNG images are downloaded directly via the browser's download mechanism.
To visualize orbitals and densities with lgv,
you have to compute a grid file (.grid)
first using the GRID_IT program from
the MOLCAS package.
Note that the quality of the picture depends on
the keywords used in GRID_IT input.
Orbitals can be browsed with the PageUp/PageDown keys, or selected by a menu, invoked by the right mouse button. If you know the symmetry and number of the orbital you want to display, you can press the F4 (or =) key and then type # followed by the symmetry and orbital number, e.g. (# 1 3).
To change the isosurface value, you can use + or - key, or press F4 key, and type the desired isosurface value.
Sometimes you may want to filter the orbitals shown by lgv. Pressing Delete key you can hide an orbital. All hidden orbitals become visible again when the Insert key is pressed. Alternatively, you can apply a filter to hide some orbitals by a criterion: symmetry number (s), orbital energy (e), occupation number (o), or typeindex (i). The use of filters is clear from the following examples: Press the F4 key and type #: followed by a filter command - #:s14 to display orbitals only from symmetry 1 and 4, #:e-2:1 to display orbitals in an energy range between -2 and 1.
When the grid file is loaded, lgv displays subspaces (frozen, inactive, RAS1, RAS2, RAS3, secondary, deleted). The user can modify the type index of the orbital, save (F2 key) the INPORB file (it will have an extension GvOrb), and use this file in a subsequent RASSCF calculation without having to reorder the orbitals. In order to modify the index of the displayed orbital, user can use a menu, or press one of the keys: fi123sd. Pressing Space key (or middle mouse button) changes the type index in a loop.
It is possible to display all orbitals of the grid file simultaneously. Press the F3 key to get the screen with all orbitals.

By default, the background (rainbow colors) for each orbital corresponds to the type index information. Clicking on an individual orbital you can use the same keys to modify it's type, or delete it from the screen. Pressing F3 button again, or Escape will close the multiview mode. Using PageUp/PageDown in multiview mode will increase/decrease the sizes of subscreens. These features of lgv can be quite helpful for selecting the different orbital spaces in RASSCF calculations.
lgv can also be used to compare densities from different GRID_IT calculations. A command lgv --minus scf.grid rasscf.grid --out diff.grid computes the density difference (SCF − RASSCF) and writes it to diff.grid. The shorthand flags --minus and --plus are equivalent to -a -1 and -a 1 respectively.
The simplest case requires both grid files to have the same mesh (same Net= dimensions). If the two calculations used the same coordinate frame but different mesh resolutions, add --same-space: lgv will trilinearly resample the first grid onto the second grid’s mesh before forming the linear combination.
If the two densities come from slightly different molecular geometries (e.g. consecutive steps of a geometry optimisation or points along an IRC path), use --align. lgv checks that both files contain the same atoms in the same order, verifies that no atom has moved more than the displacement tolerance (default 2 Å, overridden with --align-tol), and then computes an optimal rigid-body transformation (Kabsch rotation + translation) from the paired atom positions. Grid2 is resampled onto grid1’s coordinate frame using trilinear interpolation, the linear combination is formed, and the result is written on grid1’s mesh with grid1’s atom positions. The alignment RMSD is printed to stderr so you can check whether the fit is reasonable:
lgv --align --minus step_001.grid step_002.grid --out ddiff.grid
For the interaction-density workflow (comparing the density of a complex AB to the sum of its fragments A and B in ghost-atom calculations) the grids must share the same coordinate frame. Compute the fragment sum first: lgv --plus A.grid B.grid --out sum.grid, then subtract it from the complex: lgv --minus AB.grid sum.grid --out interaction.grid.
lgv can compute and display an electrostatic potential (ESP) surface: a Gaussian-blended van der Waals isosurface colored by the Coulomb potential, with red for electron-rich (negative) regions and blue for electron-poor (positive) regions. The ESP is computed as
E(r) = ∑i qi / |r − ri|
where qi are point charges at the atomic positions. Three sources of charges are supported.
If a .molden file is loaded that contains a [CHARGE] (MULLIKEN) or [CHARGE] (LOPROP) section, the ESP surface can be computed directly:
LoProp charges (if present) are preferred over Mulliken charges when both sections exist.
A plain-text file with one atom per line in the format
N comment Element x y z charge ...
where coordinates are in Ångström and charges in electron units (e), can be loaded via Right-click → ESP surface → From XYZ+charges file (native) or the Actions menu in the browser build (upload button).
A Gaussian Cube file (.cube) containing an electrostatic potential grid — as produced by Gaussian, ORCA, or similar programs — can be loaded via Right-click → ESP surface → From EPOT grid (Cube format) or the Actions menu in the browser build (upload button). The atom positions from the currently loaded file are used for the van der Waals surface; the cube grid values are interpolated onto the surface vertices by trilinear interpolation. Cube coordinates are converted from Bohr to Ångström and values from e/Bohr to e/Å automatically. The color range is auto-scaled from the cube data.
The script molden2esp_lus.py (included in the lgv distribution) reads atom positions and Mulliken/LoProp charges from a Molden file, computes the Coulomb ESP on a 3-D grid, and writes a Luscus .lus file with GridName= Electrostatic that can be loaded into lgv as a regular grid file for isosurface display:
python3 molden2esp_lus.py scf.molden esp.lus python3 molden2esp_lus.py scf.molden esp.lus --step 0.2 --pad 3.0
The --step option sets the grid spacing in Ångström (default 0.25) and --pad sets the margin beyond the van der Waals radius of each atom (default 3.0 Å). The script requires only the Python standard library.
The color mapping uses three parameters (defined in esp.c):
Use Show/Hide ESP surface to toggle visibility without discarding the computed surface, and Clear ESP surface to remove it. Loading a new file clears the ESP surface automatically.